GIPC Proteins Negatively Modulate Plexind1 Signaling During Vascular Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PLATFORM ABSTRACTS Abstract Abstract Numbers Numbers Tuesday, November 6 41
American Society of Human Genetics 62nd Annual Meeting November 6–10, 2012 San Francisco, California PLATFORM ABSTRACTS Abstract Abstract Numbers Numbers Tuesday, November 6 41. Genes Underlying Neurological Disease Room 134 #196–#204 2. 4:30–6:30pm: Plenary Abstract 42. Cancer Genetics III: Common Presentations Hall D #1–#6 Variants Ballroom 104 #205–#213 43. Genetics of Craniofacial and Wednesday, November 7 Musculoskeletal Disorders Room 124 #214–#222 10:30am–12:45 pm: Concurrent Platform Session A (11–19): 44. Tools for Phenotype Analysis Room 132 #223–#231 11. Genetics of Autism Spectrum 45. Therapy of Genetic Disorders Room 130 #232–#240 Disorders Hall D #7–#15 46. Pharmacogenetics: From Discovery 12. New Methods for Big Data Ballroom 103 #16–#24 to Implementation Room 123 #241–#249 13. Cancer Genetics I: Rare Variants Room 135 #25–#33 14. Quantitation and Measurement of Friday, November 9 Regulatory Oversight by the Cell Room 134 #34–#42 8:00am–10:15am: Concurrent Platform Session D (47–55): 15. New Loci for Obesity, Diabetes, and 47. Structural and Regulatory Genomic Related Traits Ballroom 104 #43–#51 Variation Hall D #250–#258 16. Neuromuscular Disease and 48. Neuropsychiatric Disorders Ballroom 103 #259–#267 Deafness Room 124 #52–#60 49. Common Variants, Rare Variants, 17. Chromosomes and Disease Room 132 #61–#69 and Everything in-Between Room 135 #268–#276 18. Prenatal and Perinatal Genetics Room 130 #70–#78 50. Population Genetics Genome-Wide Room 134 #277–#285 19. Vascular and Congenital Heart 51. Endless Forms Most Beautiful: Disease Room 123 #79–#87 Variant Discovery in Genomic Data Ballroom 104 #286–#294 52. -

The Extracellular Matrix Phenome Across Species
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.06.980169; this version posted March 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The extracellular matrix phenome across species Cyril Statzer1 and Collin Y. Ewald1* 1 Eidgenössische Technische Hochschule Zürich, Department of Health Sciences and Technology, Institute of Translational Medicine, Schwerzenbach-Zürich CH-8603, Switzerland *Corresponding authors: [email protected] (CYE) Keywords: Phenome, genotype-to-phenotype, matrisome, extracellular matrix, collagen, data mining. Highlights • 7.6% of the human phenome originates from variations in matrisome genes • 11’671 phenotypes are linked to matrisome genes of humans, mice, zebrafish, Drosophila, and C. elegans • Expected top ECM phenotypes are developmental, morphological and structural phenotypes • Nonobvious top ECM phenotypes include immune system, stress resilience, and age-related phenotypes 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.03.06.980169; this version posted March 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 Abstract 2 Extracellular matrices are essential for cellular and organismal function. Recent 3 genome-wide and phenome-wide association studies started to reveal a broad 4 spectrum of phenotypes associated with genetic variants. However, the phenome or 5 spectrum of all phenotypes associated with genetic variants in extracellular matrix 6 genes is unknown. -

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

Protein-Coding Variants Implicate Novel Genes Related to Lipid Homeostasis
bioRxiv preprint doi: https://doi.org/10.1101/352674; this version posted June 30, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 PROTEIN-CODING VARIANTS IMPLICATE NOVEL GENES RELATED TO LIPID HOMEOSTASIS 2 CONTRIBUTING TO BODY FAT DISTRIBUTION 3 Anne E Justice¥,1,2, Tugce Karaderi¥,3,4, Heather M Highland¥,1,5, Kristin L Young¥,1, Mariaelisa Graff¥,1, 4 Yingchang Lu¥,6,7,8, Valérie Turcot9, Paul L Auer10, Rebecca S Fine11,12,13, Xiuqing Guo14, Claudia 5 Schurmann7,8, Adelheid Lempradl15, Eirini Marouli16, Anubha Mahajan3, Thomas W Winkler17, Adam E 6 Locke18,19, Carolina Medina-Gomez20,21, Tõnu Esko11,13,22, Sailaja Vedantam11,12,13, Ayush Giri23, Ken Sin 7 Lo9, Tamuno Alfred7, Poorva Mudgal24, Maggie CY Ng24,25, Nancy L Heard-Costa26,27, Mary F Feitosa28, 8 Alisa K Manning11,29,30 , Sara M Willems31, Suthesh Sivapalaratnam30,32,33, Goncalo Abecasis18, Dewan S 9 Alam34, Matthew Allison35, Philippe Amouyel36,37,38, Zorayr Arzumanyan14, Beverley Balkau39, Lisa 10 Bastarache40, Sven Bergmann41,42, Lawrence F Bielak43, Matthias Blüher44,45, Michael Boehnke18, Heiner 11 Boeing46, Eric Boerwinkle47,48, Carsten A Böger49, Jette Bork-Jensen50, Erwin P Bottinger7, Donald W 12 Bowden24,25,51, Ivan Brandslund52,53, Linda Broer21, Amber A Burt54, Adam S Butterworth55,56, Mark J 13 Caulfield16,57, Giancarlo Cesana58, John C Chambers59,60,61,62,63, Daniel I Chasman11,64,65,66, Yii-Der Ida 14 Chen14, Rajiv Chowdhury55, Cramer Christensen67, -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Noninvasive Sleep Monitoring in Large-Scale Screening of Knock-Out Mice
bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Noninvasive sleep monitoring in large-scale screening of knock-out mice reveals novel sleep-related genes Shreyas S. Joshi1*, Mansi Sethi1*, Martin Striz1, Neil Cole2, James M. Denegre2, Jennifer Ryan2, Michael E. Lhamon3, Anuj Agarwal3, Steve Murray2, Robert E. Braun2, David W. Fardo4, Vivek Kumar2, Kevin D. Donohue3,5, Sridhar Sunderam6, Elissa J. Chesler2, Karen L. Svenson2, Bruce F. O'Hara1,3 1Dept. of Biology, University of Kentucky, Lexington, KY 40506, USA, 2The Jackson Laboratory, Bar Harbor, ME 04609, USA, 3Signal solutions, LLC, Lexington, KY 40503, USA, 4Dept. of Biostatistics, University of Kentucky, Lexington, KY 40536, USA, 5Dept. of Electrical and Computer Engineering, University of Kentucky, Lexington, KY 40506, USA. 6Dept. of Biomedical Engineering, University of Kentucky, Lexington, KY 40506, USA. *These authors contributed equally Address for correspondence and proofs: Shreyas S. Joshi, Ph.D. Dept. of Biology University of Kentucky 675 Rose Street 101 Morgan Building Lexington, KY 40506 U.S.A. Phone: (859) 257-2805 FAX: (859) 257-1717 Email: [email protected] Running title: Sleep changes in knockout mice bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
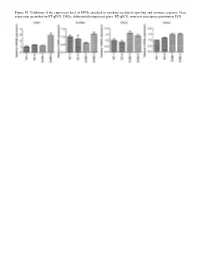
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

Review of Hair Cell Synapse Defects in Sensorineural Hearing Impairment
Otology & Neurotology 34:995Y1004 Ó 2013, Otology & Neurotology, Inc. Review of Hair Cell Synapse Defects in Sensorineural Hearing Impairment *†‡Tobias Moser, *Friederike Predoehl, and §Arnold Starr *InnerEarLab, Department of Otolaryngology, University of Go¨ttingen Medical School; ÞSensory Research Center SFB 889, þBernstein Center for Computational Neuroscience, University of Go¨ttingen, Go¨ttingen, Germany; and §Department of Neurology, University of California, Irvine, California, U.S.A. Objective: To review new insights into the pathophysiology of are similar to those accompanying auditory neuropathy, a group sensorineural hearing impairment. Specifically, we address defects of genetic and acquired disorders of spiral ganglion neurons. of the ribbon synapses between inner hair cells and spiral ganglion Genetic auditory synaptopathies include alterations of glutamate neurons that cause auditory synaptopathy. loading of synaptic vesicles, synaptic Ca2+ influx or synaptic Data Sources and Study Selection: Here, we review original vesicle turnover. Acquired synaptopathies include noise-induced publications on the genetics, animal models, and molecular hearing loss because of excitotoxic synaptic damage and subse- mechanisms of hair cell ribbon synapses and their dysfunction. quent gradual neural degeneration. Alterations of ribbon synapses Conclusion: Hair cell ribbon synapses are highly specialized to likely also contribute to age-related hearing loss. Key Words: enable indefatigable sound encoding with utmost temporal precision. GeneticsVIon -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Integrative Bulk and Single-Cell Profiling of Premanufacture T-Cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-Cell Therapy
Published OnlineFirst April 5, 2021; DOI: 10.1158/2159-8290.CD-20-1677 RESEARCH ARTICLE Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy Gregory M. Chen1, Changya Chen2,3, Rajat K. Das2, Peng Gao2, Chia-Hui Chen2, Shovik Bandyopadhyay4, Yang-Yang Ding2,5, Yasin Uzun2,3, Wenbao Yu2, Qin Zhu1, Regina M. Myers2, Stephan A. Grupp2,5, David M. Barrett2,5, and Kai Tan2,3,5 Downloaded from cancerdiscovery.aacrjournals.org on October 1, 2021. © 2021 American Association for Cancer Research. Published OnlineFirst April 5, 2021; DOI: 10.1158/2159-8290.CD-20-1677 ABSTRACT The adoptive transfer of chimeric antigen receptor (CAR) T cells represents a breakthrough in clinical oncology, yet both between- and within-patient differences in autologously derived T cells are a major contributor to therapy failure. To interrogate the molecular determinants of clinical CAR T-cell persistence, we extensively characterized the premanufacture T cells of 71 patients with B-cell malignancies on trial to receive anti-CD19 CAR T-cell therapy. We performed RNA-sequencing analysis on sorted T-cell subsets from all 71 patients, followed by paired Cellular Indexing of Transcriptomes and Epitopes (CITE) sequencing and single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) on T cells from six of these patients. We found that chronic IFN signaling regulated by IRF7 was associated with poor CAR T-cell persistence across T-cell subsets, and that the TCF7 regulon not only associates with the favorable naïve T-cell state, but is maintained in effector T cells among patients with long-term CAR T-cell persistence. -

1 Supplemental Table 1. Demographics, Clinicopathological
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut 1 Supplemental Table 1. Demographics, clinicopathological, and operative characteristics of archived iCCA specimens included in immunohistochemical survival correlative analyses. Patients with perioperative mortality (within 30 days) were excluded from survival analysis. Supplemental Table 2. Demographics, clinicopathological, and treatment characteristics of patients with unresectable CCA treated at the University of Rochester Medical Center with complete blood counts included for peripheral monocyte and neutrophil to lymphocyte ratio analysis. Univariate and Multivariate Models constructed using the Cox Proportional Hazard method [risk ratio (95% Confidence interval)]. Supplemental Table 3. Antibodies used for IHC, IHF, and flow cytometry analysis. IHC, immunohistochemistry; IHF, immunofluorescence; FC, flow cytometry. Supplemental Table 4 Enrollment characteristics of spontaneous tumour bearing mice enrolled into therapeutic trial. P value determined by Mann-Whitney U or χ2 test. Supplemental Table 5 Differentially expressed protein coding genes (DEGs) from RNA-sequencing analysis of bone marrow derived macrophages educated with tumour supernatant. DEGs compare anti-GM-CSF and IgG control treated conditions.Genes filtered to include DEGs with Log2(Fold Change) < -1.0 or >1.0 and p< 0.05. Supplemental Table 6 Pathway enrichment analysis of downregulated Gene Ontology Biological Processes from RNA- sequencing analysis of bone marrow derived macrophages educated with tumour supernatant. Pathways compare anti-GM-CSF and IgG control treated conditions. Gene sets were filtered based on p value <0.05 and Log2(Fold Change) >1.5. Supplemental Table 7 Differentially expressed protein coding genes (DEGs) from RNA-sequencing analysis of URCCA4.3 treated tumours. -

Different Species Common Arthritis
High Resolution Mapping of Cia3: A Common Arthritis Quantitative Trait Loci in Different Species This information is current as Xinhua Yu, Haidong Teng, Andreia Marques, Farahnaz of September 27, 2021. Ashgari and Saleh M. Ibrahim J Immunol 2009; 182:3016-3023; ; doi: 10.4049/jimmunol.0803005 http://www.jimmunol.org/content/182/5/3016 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2009/02/18/182.5.3016.DC1 Material References This article cites 36 articles, 9 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/182/5/3016.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology High Resolution Mapping of Cia3: A Common Arthritis Quantitative Trait Loci in Different Species1 Xinhua Yu, Haidong Teng, Andreia Marques, Farahnaz Ashgari, and Saleh M.