RNA-Seq Analysis Pipeline for Genomics Scientists
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Efficacy and Mechanistic Evaluation of Tic10, a Novel Antitumor Agent
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2012 Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Joshua Edward Allen University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Oncology Commons Recommended Citation Allen, Joshua Edward, "Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent" (2012). Publicly Accessible Penn Dissertations. 488. https://repository.upenn.edu/edissertations/488 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/488 For more information, please contact [email protected]. Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Abstract TNF-related apoptosis-inducing ligand (TRAIL; Apo2L) is an endogenous protein that selectively induces apoptosis in cancer cells and is a critical effector in the immune surveillance of cancer. Recombinant TRAIL and TRAIL-agonist antibodies are in clinical trials for the treatment of solid malignancies due to the cancer-specific cytotoxicity of TRAIL. Recombinant TRAIL has a short serum half-life and both recombinant TRAIL and TRAIL receptor agonist antibodies have a limited capacity to perfuse to tissue compartments such as the brain, limiting their efficacy in certain malignancies. To overcome such limitations, we searched for small molecules capable of inducing the TRAIL gene using a high throughput luciferase reporter gene assay. We selected TRAIL-inducing compound 10 (TIC10) for further study based on its induction of TRAIL at the cell surface and its promising therapeutic index. TIC10 is a potent, stable, and orally active antitumor agent that crosses the blood-brain barrier and transcriptionally induces TRAIL and TRAIL-mediated cell death in a p53-independent manner. -

The Expression of the Human Apolipoprotein Genes and Their Regulation by Ppars
CORE Metadata, citation and similar papers at core.ac.uk Provided by UEF Electronic Publications The expression of the human apolipoprotein genes and their regulation by PPARs Juuso Uski M.Sc. Thesis Biochemistry Department of Biosciences University of Kuopio June 2008 Abstract The expression of the human apolipoprotein genes and their regulation by PPARs. UNIVERSITY OF KUOPIO, the Faculty of Natural and Environmental Sciences, Curriculum of Biochemistry USKI Juuso Oskari Thesis for Master of Science degree Supervisors Prof. Carsten Carlberg, Ph.D. Merja Heinäniemi, Ph.D. June 2008 Keywords: nuclear receptors; peroxisome proliferator-activated receptor; PPAR response element; apolipoprotein; lipid metabolism; high density lipoprotein; low density lipoprotein. Lipids are any fat-soluble, naturally-occurring molecules and one of their main biological functions is energy storage. Lipoproteins carry hydrophobic lipids in the water and salt-based blood environment for processing and energy supply in liver and other organs. In this study, the genomic area around the apolipoprotein genes was scanned in silico for PPAR response elements (PPREs) using the in vitro data-based computer program. Several new putative REs were found in surroundings of multiple lipoprotein genes. The responsiveness of those apolipoprotein genes to the PPAR ligands GW501516, rosiglitazone and GW7647 in the HepG2, HEK293 and THP-1 cell lines were tested with real-time PCR. The APOA1, APOA2, APOB, APOD, APOE, APOF, APOL1, APOL3, APOL5 and APOL6 genes were found to be regulated by PPARs in direct or secondary manners. Those results provide new insights in the understanding of lipid metabolism and so many lifestyle diseases like atherosclerosis, type 2 diabetes, heart disease and stroke. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Pdf%5Cn T/32/Suppl 1/D267.Short%5Cn
University of Kentucky UKnowledge Theses and Dissertations--Molecular and Cellular Biochemistry Molecular and Cellular Biochemistry 2019 COMPUTATIONAL TOOLS FOR THE DYNAMIC CATEGORIZATION AND AUGMENTED UTILIZATION OF THE GENE ONTOLOGY Eugene Waverly Hinderer III University of Kentucky, [email protected] Author ORCID Identifier: https://orcid.org/0000-0001-8045-7083 Digital Object Identifier: https://doi.org/10.13023/etd.2019.362 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Hinderer, Eugene Waverly III, "COMPUTATIONAL TOOLS FOR THE DYNAMIC CATEGORIZATION AND AUGMENTED UTILIZATION OF THE GENE ONTOLOGY" (2019). Theses and Dissertations--Molecular and Cellular Biochemistry. 43. https://uknowledge.uky.edu/biochem_etds/43 This Doctoral Dissertation is brought to you for free and open access by the Molecular and Cellular Biochemistry at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Molecular and Cellular Biochemistry by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

4E-BP2 / EIF4EBP2 Antibody (Aa99-120) Rabbit Polyclonal Antibody Catalog # ALS12947
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 4E-BP2 / EIF4EBP2 Antibody (aa99-120) Rabbit Polyclonal Antibody Catalog # ALS12947 Specification 4E-BP2 / EIF4EBP2 Antibody (aa99-120) - Product Information Application IHC Primary Accession Q13542 Reactivity Human, Mouse, Rat, Pig, Bovine, Dog Host Rabbit Clonality Polyclonal Calculated MW 13kDa KDa 4E-BP2 / EIF4EBP2 Antibody (aa99-120) - Additional Information Anti-EIF4EBP2 / 4EBP2 antibody IHC of Gene ID 1979 human pancreas. Other Names Eukaryotic translation initiation factor 4E-BP2 / EIF4EBP2 Antibody (aa99-120) - 4E-binding protein 2, 4E-BP2, eIF4E-binding Background protein 2, EIF4EBP2 Regulates eIF4E activity by preventing its Target/Specificity assembly into the eIF4F complex. Mediates the Detects 17 and 20 kD proteins, regulation of protein translation by hormones, corresponding to the apparent molecular growth factors and other stimuli that signal mass of PHAS-II and its phosphorylated through the MAP kinase pathway. state on SDS-PAGE immunoblots. 4E-BP2 / EIF4EBP2 Antibody (aa99-120) - Reconstitution & Storage References Short term 4°C, long term aliquot and store at -20°C, avoid freeze thaw cycles. For Pause A.,et al.Nature 371:762-767(1994). maximum product recovery, after thawing, Kalnine N.,et al.Submitted (MAY-2003) to the centrifuge the product vial before removing cap. EMBL/GenBank/DDBJ databases. Dephoure N.,et al.Proc. Natl. Acad. Sci. U.S.A. Precautions 105:10762-10767(2008). 4E-BP2 / EIF4EBP2 Antibody (aa99-120) is Gauci S.,et al.Anal. Chem. for research use only and not for use in 81:4493-4501(2009). diagnostic or therapeutic procedures. -
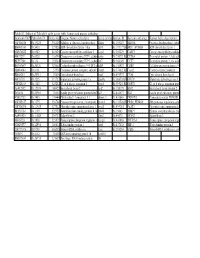
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

Cacybp (NM 009786) Mouse Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for MR202748 Cacybp (NM_009786) Mouse Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Cacybp (NM_009786) Mouse Tagged ORF Clone Tag: Myc-DDK Symbol: Cacybp Synonyms: SIP Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >MR202748 ORF sequence Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCTTCCGTTTTGGAAGAGTTGCAGAAAGACCTAGAAGAGGTCAAAGTATTGCTGGAAAAGTCCACTA GGAAAAGACTACGTGATACTCTTACAAGTGAAAAGTCCAAGATTGAGACGGAACTCAAGAACAAGATGCA ACAGAAGTCGCAGAAGAAACCAGAACTTGATAATGAAAAGCCAGCTGCTGTGGTTGCTCCTCTTACAACA GGATACACCGTGAAAATCAGTAATTATGGATGGGATCAGTCAGATAAGTTTGTGAAAATCTACATTACCT TGACTGGCGTCCATCAGGTGCCCACTGAGAACGTGCAGGTGCACTTCACAGAGAGGTCATTTGATCTTCT GGTAAAAAACCTCAATGGCAAGAATTACTCCATGATTGTGAACAATCTTTTGAAACCTATCTCTGTGGAA AGCAGTTCAAAAAAAGTCAAGACTGATACAGTAATTATTCTATGTAGAAAGAAAGCAGAAAACACAAGAT GGGACTACTTAACACAGGTGGAAAAGGAATGCAAAGAGAAAGAAAAGCCTTCCTACGACACGGAGGCAGA CCCTAGTGAGGGATTAATGAATGTTCTAAAGAAAATTTATGAAGACGGAGACGATGATATGAAGCGAACC ATTAATAAAGCGTGGGTGGAATCCAGAGAGAAGCAAGCCAGAGAAGACACGGAATTT ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT ACAAGGATGACGACGATAAGGTTTAA This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

CCT6A Sirna (Mouse)
For research purposes only, not for human use Product Data Sheet CCT6A siRNA (Mouse) Catalog # Source Reactivity Applications CRM0566 Synthetic M RNAi Description siRNA to inhibit CCT6A expression using RNA interference Specificity CCT6A siRNA (Mouse) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol CCT6A Alternative Names CCT6; CCTZ; CCTZ1; T-complex protein 1 subunit zeta; TCP-1-zeta; CCT-zeta-1 Entrez Gene 12466 (Mouse) SwissProt P80317 (Mouse) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of mouse CCT6A gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds.