75 2. INTRODUCTION Triple-Negative Breast Cancer (TNBC)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
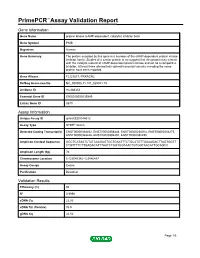
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name protein kinase (cAMP-dependent, catalytic) inhibitor beta Gene Symbol PKIB Organism Human Gene Summary The protein encoded by this gene is a member of the cAMP-dependent protein kinase inhibitor family. Studies of a similar protein in rat suggest that this protein may interact with the catalytic subunit of cAMP-dependent protein kinase and act as a competitive inhibitor. At least three alternatively spliced transcript variants encoding the same protein have been reported. Gene Aliases FLJ23817, PRKACN2 RefSeq Accession No. NC_000006.11, NT_025741.15 UniGene ID Hs.486354 Ensembl Gene ID ENSG00000135549 Entrez Gene ID 5570 Assay Information Unique Assay ID qHsaCED0044612 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000368452, ENST00000368448, ENST00000258014, ENST00000354275, ENST00000368446, ENST00000392491, ENST00000392490 Amplicon Context Sequence GGCTCATAATCTATCAAGAGTGCTGAATTTCTGCATGTTGAAAGACTTAGTGGTT CTGTTTTCTTGAGACATTTAATCTGGTGGTAACTGTGGTAACATTGCAGCC Amplicon Length (bp) 76 Chromosome Location 6:123046342-123046447 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9996 cDNA Cq 22.83 cDNA Tm (Celsius) 76.5 gDNA Cq 24.52 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report PKIB, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve -

Location Analysis of Estrogen Receptor Target Promoters Reveals That
Location analysis of estrogen receptor ␣ target promoters reveals that FOXA1 defines a domain of the estrogen response Jose´ e Laganie` re*†, Genevie` ve Deblois*, Ce´ line Lefebvre*, Alain R. Bataille‡, Franc¸ois Robert‡, and Vincent Gigue` re*†§ *Molecular Oncology Group, Departments of Medicine and Oncology, McGill University Health Centre, Montreal, QC, Canada H3A 1A1; †Department of Biochemistry, McGill University, Montreal, QC, Canada H3G 1Y6; and ‡Laboratory of Chromatin and Genomic Expression, Institut de Recherches Cliniques de Montre´al, Montreal, QC, Canada H2W 1R7 Communicated by Ronald M. Evans, The Salk Institute for Biological Studies, La Jolla, CA, July 1, 2005 (received for review June 3, 2005) Nuclear receptors can activate diverse biological pathways within general absence of large scale functional data linking these putative a target cell in response to their cognate ligands, but how this binding sites with gene expression in specific cell types. compartmentalization is achieved at the level of gene regulation is Recently, chromatin immunoprecipitation (ChIP) has been used poorly understood. We used a genome-wide analysis of promoter in combination with promoter or genomic DNA microarrays to occupancy by the estrogen receptor ␣ (ER␣) in MCF-7 cells to identify loci recognized by transcription factors in a genome-wide investigate the molecular mechanisms underlying the action of manner in mammalian cells (20–24). This technology, termed 17-estradiol (E2) in controlling the growth of breast cancer cells. ChIP-on-chip or location analysis, can therefore be used to deter- We identified 153 promoters bound by ER␣ in the presence of E2. mine the global gene expression program that characterize the Motif-finding algorithms demonstrated that the estrogen re- action of a nuclear receptor in response to its natural ligand. -

Silkworm Z Chromosome Is Enriched in Testis-Specific Genes
Supporting Information http://www.genetics.org/cgi/content/full/genetics.108.099994/DC1 Silkworm Z Chromosome is Enriched in Testis-Specific Genes K. P. Arunkumar, Kazuei Mita and J. Nagaraju Copyright © 2009 by the Genetics Society of America DOI: 10.1534/genetics.108.099994 2 SI K. Arunkumar et al. File S1 Gene Ontology annotation GO annotation generates a dynamic controlled vocabulary that can be applied to all organisms, even while knowledge of gene and protein roles in cells is still accumulating and changing. To this end, the Seqdblite FASTA sequence flat file was downloaded from the GO database. By running BLAST against Seqdblite, closest homologue was identified. From BLAST output, molecular functions, biological processes and cellular localisation were parsed by building an in-house GO database in MySQL from the GO-term-database flat file, downloaded from Gene Ontology Database Downloads (http://www.godatabase.org/dev/). The Perl-DBI was used to interface with MySQL, to extract the parent terms of each individual GO term that are obtained by parsing BLAST output. The output was then represented graphically. All ESTs were assigned a biological process, molecular function and cellular component using Gene Ontology (GO) database. The closest annotated homologue in the GO database was used for assigning these categories. The results of the GO annotation are graphically represented in Figures S1-3. Many of the gene products were found to be localized in cell (42%). In cell, gene products were predominant in intracellular region (78%) which comprised of localizations in intracellular organelle (38%) and cytoplasm (29%). The other localizations were organelle (29%) followed by protein complex (18%) (Figure S1). -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Molecular Effects of Isoflavone Supplementation Human Intervention Studies and Quantitative Models for Risk Assessment
Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis committee Promotors Prof. Dr Pieter van ‘t Veer Professor of Nutritional Epidemiology Wageningen University Prof. Dr Evert G. Schouten Emeritus Professor of Epidemiology and Prevention Wageningen University Co-promotors Dr Anouk Geelen Assistant professor, Division of Human Nutrition Wageningen University Dr Lydia A. Afman Assistant professor, Division of Human Nutrition Wageningen University Other members Prof. Dr Jaap Keijer, Wageningen University Dr Hubert P.J.M. Noteborn, Netherlands Food en Consumer Product Safety Authority Prof. Dr Yvonne T. van der Schouw, UMC Utrecht Dr Wendy L. Hall, King’s College London This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences). Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 20 June 2014 at 13.30 p.m. in the Aula. Vera van der Velpen Molecular effects of isoflavone supplementation: Human intervention studies and quantitative models for risk assessment 154 pages PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN: 978-94-6173-952-0 ABSTRact Background: Risk assessment can potentially be improved by closely linked experiments in the disciplines of epidemiology and toxicology. -

Proteome-Wide Survey of the Autoimmune Target Repertoire In
www.nature.com/scientificreports OPEN Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine Received: 28 October 2015 Accepted: 23 December 2015 syndrome type 1 Published: 01 February 2016 Nils Landegren1,2,*, Donald Sharon3,4,*, Eva Freyhult2,5,6, Åsa Hallgren1,2, Daniel Eriksson1,2, Per-Henrik Edqvist7, Sophie Bensing8, Jeanette Wahlberg9, Lawrence M. Nelson10, Jan Gustafsson11, Eystein S Husebye12, Mark S Anderson13, Michael Snyder3 & Olle Kämpe1,2 Autoimmune polyendocrine syndrome type 1 (APS1) is a monogenic disorder that features multiple autoimmune disease manifestations. It is caused by mutations in the Autoimmune regulator (AIRE) gene, which promote thymic display of thousands of peripheral tissue antigens in a process critical for establishing central immune tolerance. We here used proteome arrays to perform a comprehensive study of autoimmune targets in APS1. Interrogation of established autoantigens revealed highly reliable detection of autoantibodies, and by exploring the full panel of more than 9000 proteins we further identified MAGEB2 and PDILT as novel major autoantigens in APS1. Our proteome-wide assessment revealed a marked enrichment for tissue-specific immune targets, mirroringAIRE ’s selectiveness for this category of genes. Our findings also suggest that only a very limited portion of the proteome becomes targeted by the immune system in APS1, which contrasts the broad defect of thymic presentation associated with AIRE-deficiency and raises novel questions what other factors are needed for break of tolerance. Autoimmune responses can ultimately be defined at the molecular level by the specific interaction between T- or B-cell receptors and a distinct self-molecule. In tissue-specific autoimmune disorders the immune system typically target molecules that are exclusively expressed in the affected tissue and involve a combined cellular and humoral response with cognate specificities1–3. -

The Correlation of Keratin Expression with In-Vitro Epithelial Cell Line Differentiation
The correlation of keratin expression with in-vitro epithelial cell line differentiation Deeqo Aden Thesis submitted to the University of London for Degree of Master of Philosophy (MPhil) Supervisors: Professor Ian. C. Mackenzie Professor Farida Fortune Centre for Clinical and Diagnostic Oral Science Barts and The London School of Medicine and Dentistry Queen Mary, University of London 2009 Contents Content pages ……………………………………………………………………......2 Abstract………………………………………………………………………….........6 Acknowledgements and Declaration……………………………………………...…7 List of Figures…………………………………………………………………………8 List of Tables………………………………………………………………………...12 Abbreviations….………………………………………………………………..…...14 Chapter 1: Literature review 16 1.1 Structure and function of the Oral Mucosa……………..…………….…..............17 1.2 Maintenance of the oral cavity...……………………………………….................20 1.2.1 Environmental Factors which damage the Oral Mucosa………. ….…………..21 1.3 Structure and function of the Oral Mucosa ………………...….……….………...21 1.3.1 Skin Barrier Formation………………………………………………….……...22 1.4 Comparison of Oral Mucosa and Skin…………………………………….……...24 1.5 Developmental and Experimental Models used in Oral mucosa and Skin...……..28 1.6 Keratinocytes…………………………………………………….….....................29 1.6.1 Desmosomes…………………………………………….…...............................29 1.6.2 Hemidesmosomes……………………………………….…...............................30 1.6.3 Tight Junctions………………………….……………….…...............................32 1.6.4 Gap Junctions………………………….……………….….................................32 -

Proteomic Expression Profile in Human Temporomandibular Joint
diagnostics Article Proteomic Expression Profile in Human Temporomandibular Joint Dysfunction Andrea Duarte Doetzer 1,*, Roberto Hirochi Herai 1 , Marília Afonso Rabelo Buzalaf 2 and Paula Cristina Trevilatto 1 1 Graduate Program in Health Sciences, School of Medicine, Pontifícia Universidade Católica do Paraná (PUCPR), Curitiba 80215-901, Brazil; [email protected] (R.H.H.); [email protected] (P.C.T.) 2 Department of Biological Sciences, Bauru School of Dentistry, University of São Paulo, Bauru 17012-901, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-41-991-864-747 Abstract: Temporomandibular joint dysfunction (TMD) is a multifactorial condition that impairs human’s health and quality of life. Its etiology is still a challenge due to its complex development and the great number of different conditions it comprises. One of the most common forms of TMD is anterior disc displacement without reduction (DDWoR) and other TMDs with distinct origins are condylar hyperplasia (CH) and mandibular dislocation (MD). Thus, the aim of this study is to identify the protein expression profile of synovial fluid and the temporomandibular joint disc of patients diagnosed with DDWoR, CH and MD. Synovial fluid and a fraction of the temporomandibular joint disc were collected from nine patients diagnosed with DDWoR (n = 3), CH (n = 4) and MD (n = 2). Samples were subjected to label-free nLC-MS/MS for proteomic data extraction, and then bioinformatics analysis were conducted for protein identification and functional annotation. The three Citation: Doetzer, A.D.; Herai, R.H.; TMD conditions showed different protein expression profiles, and novel proteins were identified Buzalaf, M.A.R.; Trevilatto, P.C. -

MALE Protein Name Accession Number Molecular Weight CP1 CP2 H1 H2 PDAC1 PDAC2 CP Mean H Mean PDAC Mean T-Test PDAC Vs. H T-Test
MALE t-test t-test Accession Molecular H PDAC PDAC vs. PDAC vs. Protein Name Number Weight CP1 CP2 H1 H2 PDAC1 PDAC2 CP Mean Mean Mean H CP PDAC/H PDAC/CP - 22 kDa protein IPI00219910 22 kDa 7 5 4 8 1 0 6 6 1 0.1126 0.0456 0.1 0.1 - Cold agglutinin FS-1 L-chain (Fragment) IPI00827773 12 kDa 32 39 34 26 53 57 36 30 55 0.0309 0.0388 1.8 1.5 - HRV Fab 027-VL (Fragment) IPI00827643 12 kDa 4 6 0 0 0 0 5 0 0 - 0.0574 - 0.0 - REV25-2 (Fragment) IPI00816794 15 kDa 8 12 5 7 8 9 10 6 8 0.2225 0.3844 1.3 0.8 A1BG Alpha-1B-glycoprotein precursor IPI00022895 54 kDa 115 109 106 112 111 100 112 109 105 0.6497 0.4138 1.0 0.9 A2M Alpha-2-macroglobulin precursor IPI00478003 163 kDa 62 63 86 72 14 18 63 79 16 0.0120 0.0019 0.2 0.3 ABCB1 Multidrug resistance protein 1 IPI00027481 141 kDa 41 46 23 26 52 64 43 25 58 0.0355 0.1660 2.4 1.3 ABHD14B Isoform 1 of Abhydrolase domain-containing proteinIPI00063827 14B 22 kDa 19 15 19 17 15 9 17 18 12 0.2502 0.3306 0.7 0.7 ABP1 Isoform 1 of Amiloride-sensitive amine oxidase [copper-containing]IPI00020982 precursor85 kDa 1 5 8 8 0 0 3 8 0 0.0001 0.2445 0.0 0.0 ACAN aggrecan isoform 2 precursor IPI00027377 250 kDa 38 30 17 28 34 24 34 22 29 0.4877 0.5109 1.3 0.8 ACE Isoform Somatic-1 of Angiotensin-converting enzyme, somaticIPI00437751 isoform precursor150 kDa 48 34 67 56 28 38 41 61 33 0.0600 0.4301 0.5 0.8 ACE2 Isoform 1 of Angiotensin-converting enzyme 2 precursorIPI00465187 92 kDa 11 16 20 30 4 5 13 25 5 0.0557 0.0847 0.2 0.4 ACO1 Cytoplasmic aconitate hydratase IPI00008485 98 kDa 2 2 0 0 0 0 2 0 0 - 0.0081 - 0.0 -

Keratin 12-Deficient Mice Have Fragile Corneal Epithelia
Keratin 12-Deficient Mice Have Fragile Corneal Epithelia Winston W.—Y. Kao,* Chia-YangLiu,* Richard L. Converse,* Atsushi Shiraishi* Candace W.-C. Kao,* Masamichi Ishizaki* Thomas Doetschman,^ and John Duffy-f Purpose. Expression of the K3-K12 keratin pair characterizes the corneal epithelial differentia- tion. To elucidate the role of keratin 12 in the maintenance of corneal epithelium integrity, the authors bred mice deficient in keratin 12 by gene-targeting techniques. Methods. One allele of murine Krtl.12 gene was ablated in the embryonic stem cell line, E14.1, by homologous recombination with a DNA construct in which the DNA element between intron 2 and exon 8 of the keratin 12 gene was replaced by a neo-gene. The homologous recombinant embryonic stem cells were injected to mouse blastocysts, and germ lines of chimeras were obtained. The corneas of heterozygous and homozygous mice were characterized by clinical observations using stereomicroscopy, histology with light and electron microscopy, Western immunoblot analysis, immunohistochemistry, in situ hybridization, and Northern hybridization. Results. The heterozygous mice (+/—) one allele of the Krtl.12 gene appear normal and do not develop any clinical manifestations (e.g., corneal epithelial defects). Homozygous mice (—/—) develop normally and suffer mild corneal epithelial erosion. Their corneal epithelia are fragile and can be removed by gentle rubbing of the eyes or brushing with a Microsponge. The corneal epithelium of the homozygote (—/—) does not express keratin 12 as judged by immunohistochemis- try, Western immunoblot analysis with epitope-specific anti-keratin 12 antibodies, Nordiern hybrid- ization with 32P-labeled keratin 12 cDNA, and in situ hybridization with an anti-sense keratin 12 riboprobe. -

Integrated Analysis of Gene Expression Changes Associated
Miao et al. Lipids in Health and Disease (2019) 18:92 https://doi.org/10.1186/s12944-019-1032-5 RESEARCH Open Access Integrated analysis of gene expression changes associated with coronary artery disease Liu Miao1 , Rui-Xing Yin1,2,3* , Feng Huang1,2,3, Shuo Yang1, Wu-Xian Chen1 and Jin-Zhen Wu1 Abstract Background: This study investigated the pathways and genes involved in coronary artery disease (CAD) and the associated mechanisms. Methods: Two array data sets of GSE19339 and GSE56885 were downloaded. The limma package was used to analyze the differentially expressed genes (DEGs) in normal and CAD specimens. Examination of DEGs through Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment and Gene Ontology annotation was achieved by Database for Annotation, Visualization and Integrated Discovery (DAVID). The Cytoscape software facilitated the establishment of the protein-protein interaction (PPI) network and Molecular Complex Detection (MCODE) was performed for the significant modules. Results: We identified 413 DEGs (291 up-regulated and 122 down-regulated). Approximately 256 biological processes, only 1 cellular component, and 21 molecular functions were identified by GO analysis and 10 pathways were enriched by KEGG. Moreover, 264 protein pairs and 64 nodes were visualized by the PPI network. After the MCODE analysis, the top 4 high degree genes, including interleukin 1 beta (IL1B, degree = 29), intercellular adhesion molecule 1 (ICAM1, degree = 25), Jun proto-oncogene (JUN, degree = 23) and C-C motif chemokine ligand 2 (CCL2, degree = 20) had been identified to validate in RT-PCR and Cox proportional hazards regression between CAD and normals. Conclusions: The relative expression of IL1B, ICAM1 and CCL2 was higher in CAD than in normal controls (P <0.05–0.