What Are Their Roles in Mitochondrial Protein Synthesis?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rise and Fall of the Bovine Corpus Luteum
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Rise and Fall of the Bovine Corpus Luteum Heather Talbott University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, Molecular Biology Commons, and the Obstetrics and Gynecology Commons Recommended Citation Talbott, Heather, "The Rise and Fall of the Bovine Corpus Luteum" (2017). Theses & Dissertations. 207. https://digitalcommons.unmc.edu/etd/207 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE RISE AND FALL OF THE BOVINE CORPUS LUTEUM by Heather Talbott A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor John S. Davis University of Nebraska Medical Center Omaha, Nebraska May, 2017 Supervisory Committee: Carol A. Casey, Ph.D. Andrea S. Cupp, Ph.D. Parmender P. Mehta, Ph.D. Justin L. Mott, Ph.D. i ACKNOWLEDGEMENTS This dissertation was supported by the Agriculture and Food Research Initiative from the USDA National Institute of Food and Agriculture (NIFA) Pre-doctoral award; University of Nebraska Medical Center Graduate Student Assistantship; University of Nebraska Medical Center Exceptional Incoming Graduate Student Award; the VA Nebraska-Western Iowa Health Care System Department of Veterans Affairs; and The Olson Center for Women’s Health, Department of Obstetrics and Gynecology, Nebraska Medical Center. -

Analysis of the Relationship Between Ribosomal Protein and SSU Processome Assembly in Saccharomyces Cerevisiae
Analysis of the relationship between ribosomal protein and SSU processome assembly in Saccharomyces cerevisiae Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) der naturwissenschaftlichen Fakultät III – Biologie und vorklinische Medizin - der Universität Regensburg vorgelegt von Steffen Jakob aus Wolfen Januar 2010 Promotionsgesuch eingereicht am: 13. Januar 2010 Die Arbeit wurde angeleitet von: Prof. Dr. Herbert Tschochner Prüfungsausschuss: Vorsitzender: Prof. Dr. Armin Kurtz 1. Prüfer: Prof. Dr. Herbert Tschochner 2. Prüfer: Prof. Dr. Rainer Deutzmann 3. Prüfer: Prof. Dr. Wolfgang Seufert Tag der mündlichen Prüfung: 24. März 2010 Die vorliegende Arbeit wurde in der Zeit von April 2006 bis Januar 2010 am Lehrstuhl Biochemie III des Institutes für Biochemie, Genetik und Mikrobiologie der Naturwissenschaftlichen Fakultät III der Universität zu Regensburg unter Anleitung von Dr. Philipp Milkereit im Labor von Prof. Dr. Herbert Tschochner angefertigt. Ich erkläre hiermit, dass ich diese Arbeit selbst verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel verwendet habe. Diese Arbeit war bisher noch nicht Bestandteil eines Prüfungsverfahrens. Andere Promotionsversuche wurden nicht unternommen. Regensburg, den 13. Januar 2010 Steffen Jakob Table of Contents Table of Contents 1 SUMMARY ...................................................................................................... 1 2 INTRODUCTION ............................................................................................ -
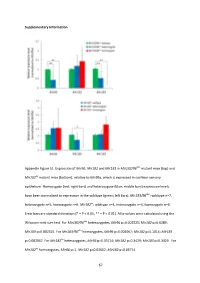
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Evaluation of Cancer-Derived Myocardial Impairments Using a Mouse Model
www.oncotarget.com Oncotarget, 2020, Vol. 11, (No. 41), pp: 3712-3722 Research Paper Evaluation of cancer-derived myocardial impairments using a mouse model Yoshihiro Miyagawa1, Shota Nukaga1,2, Takuya Mori1, Rina Fujiwara-Tani1, Kiyomu Fujii1, Shiori Mori1, Kei Goto1,3, Shingo Kishi1, Takamitsu Sasaki1, Chie Nakashima1, Hitoshi Ohmori1, Isao Kawahara1,2, Yi Luo4 and Hiroki Kuniyasu1 1Department of Molecular Pathology, Nara Medical University, Kashihara, Nara 634-8521, Japan 2Division of Rehabilitation, Hanna Central Hospital, Ikoma, Nara 630-0243, Japan 3Division of Rehabilitation, Hoshida Minami Hospital, Katano, Osaka 576-0022, Japan 4Key Laboratory of Neuroregeneration of Jiangsu and Ministry of Education, Co-Innovation Center of Neuroregeneration, Nantong University, Nantong, Jiangsu Province 226001, China Correspondence to: Yi Luo, email: [email protected] Hiroki Kuniyasu, email: [email protected] Keywords: cachexia; myocardium; atrophy; mitochondria; oxidative stress Received: June 26, 2020 Accepted: September 10, 2020 Published: October 13, 2020 Copyright: © 2020 Miyagawa et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Myocardial damage in cancer patients is emphasized as a cause of death; however, there are not many murine cachexia models to evaluate cancer-derived heart disorder. Using the mouse cachexia model that we established previously, we investigated myocardial damage in tumor-bearing mice. In cachexic mice, decreased heart weight and myocardial volume, and dilated left ventricular lumen, and atrophied cardiomyocytes were noted. The cardiomyocytes also showed accumulated 8-hydroxydeoxyguanosine, decreased leucine zipper and EF-hand- containing transmembrane protein-1, and increased microtubule-associated protein light chain3-II. -

Micrornas Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins
cells Review microRNAs Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins Abu Musa Md Talimur Reza 1,2 and Yu-Guo Yuan 1,3,* 1 Jiangsu Co-Innovation Center of Prevention and Control of Important Animal Infectious Diseases and Zoonoses, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China; [email protected] 2 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawi´nskiego5a, 02-106 Warsaw, Poland 3 Jiangsu Key Laboratory of Zoonosis/Joint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China * Correspondence: [email protected]; Tel.: +86-514-8797-9228 Abstract: Ribosomal proteins (RPs) are mostly derived from the energy-consuming enzyme families such as ATP-dependent RNA helicases, AAA-ATPases, GTPases and kinases, and are important structural components of the ribosome, which is a supramolecular ribonucleoprotein complex, composed of Ribosomal RNA (rRNA) and RPs, coordinates the translation and synthesis of proteins with the help of transfer RNA (tRNA) and other factors. Not all RPs are indispensable; in other words, the ribosome could be functional and could continue the translation of proteins instead of lacking in some of the RPs. However, the lack of many RPs could result in severe defects in the biogenesis of ribosomes, which could directly influence the overall translation processes and global expression of the proteins leading to the emergence of different diseases including cancer. While microRNAs (miRNAs) are small non-coding RNAs and one of the potent regulators of the post-transcriptional 0 gene expression, miRNAs regulate gene expression by targeting the 3 untranslated region and/or coding region of the messenger RNAs (mRNAs), and by interacting with the 50 untranslated region, Citation: Reza, A.M.M.T.; Yuan, Y.-G. -

LETM1 Gene Leucine Zipper and EF-Hand Containing Transmembrane Protein 1
LETM1 gene leucine zipper and EF-hand containing transmembrane protein 1 Normal Function The LETM1 gene provides instructions for making a protein whose function is not well understood. This protein is active in mitochondria, which are structures within cells that convert the energy from food into a form that cells can use. The LETM1 protein may be involved in the transport of charged calcium atoms (calcium ions) across membranes within mitochondria. Researchers suspect that the protein also plays a role in determining the shape and volume of mitochondria. Health Conditions Related to Genetic Changes Wolf-Hirschhorn syndrome The LETM1 gene is located in a region of chromosome 4 that is deleted in people with the typical features of Wolf-Hirschhorn syndrome. As a result of this deletion, affected individuals are missing one copy of the LETM1 gene in each cell. Studies suggest that a loss of this gene alters the structure of mitochondria; however, it is unclear how this abnormality is related to the signs and symptoms of Wolf-Hirschhorn syndrome. Specifically, a loss of the LETM1 gene has been associated with seizures or other abnormal electrical activity in the brain. Other Names for This Gene • LETM1_HUMAN • leucine zipper-EF-hand containing transmembrane protein 1 Additional Information & Resources Tests Listed in the Genetic Testing Registry • Tests of LETM1 (https://www.ncbi.nlm.nih.gov/gtr/all/tests/?term=3954[geneid]) Scientific Articles on PubMed Reprinted from MedlinePlus Genetics (https://medlineplus.gov/genetics/) 1 • PubMed (https://pubmed.ncbi.nlm.nih.gov/?term=%28%28LETM1%5BTIAB%5D%2 -

History of the Ribosome and the Origin of Translation
History of the ribosome and the origin of translation Anton S. Petrova,1, Burak Gulena, Ashlyn M. Norrisa, Nicholas A. Kovacsa, Chad R. Berniera, Kathryn A. Laniera, George E. Foxb, Stephen C. Harveyc, Roger M. Wartellc, Nicholas V. Huda, and Loren Dean Williamsa,1 aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332; bDepartment of Biology and Biochemistry, University of Houston, Houston, TX, 77204; and cSchool of Biology, Georgia Institute of Technology, Atlanta, GA 30332 Edited by David M. Hillis, The University of Texas at Austin, Austin, TX, and approved November 6, 2015 (received for review May 18, 2015) We present a molecular-level model for the origin and evolution of building up of the functional centers, proceeds to the establishment the translation system, using a 3D comparative method. In this model, of the common core, and continues to the development of large the ribosome evolved by accretion, recursively adding expansion metazoan rRNAs. segments, iteratively growing, subsuming, and freezing the rRNA. Incremental evolution of function is mapped out by stepwise Functions of expansion segments in the ancestral ribosome are accretion of rRNA. In the extant ribosome, specific segments of assigned by correspondence with their functions in the extant rRNA perform specific functions including peptidyl transfer, ribosome. The model explains the evolution of the large ribosomal subunit association, decoding, and energy-driven translocation subunit, the small ribosomal subunit, tRNA, and mRNA. Prokaryotic (11). The model assumes that the correlations of rRNA segments ribosomes evolved in six phases, sequentially acquiring capabilities with their functions have been reasonably maintained over the for RNA folding, catalysis, subunit association, correlated evolution, broad course of ribosomal evolution. -

HUMAN RIBOSOME BIOGENESIS and the REGULATION of the TUMOUR SUPPRESSOR P53
HUMAN RIBOSOME BIOGENESIS AND THE REGULATION OF THE TUMOUR SUPPRESSOR p53 Andria Pelava Submitted for Doctor of Philosophy Final submission: December 2016 Institute of Cell and Molecular Biosciences Faculty of Medical Sciences Newcastle University ii Abstract Ribosome production is an energetically expensive and, therefore, highly regulated process. Defects in ribosome biogenesis lead to genetic diseases called Ribosomopathies, such as Dyskeratosis Congenita (DC), and mutations in ribosomal proteins and ribosome biogenesis factors are linked to multiple types of cancer. During ribosome biogenesis, the ribosomal RNAs (rRNAs) are processed and modified, and defects in ribosome biogenesis lead to the activation of p53. This project aimed to investigate the functions of Dyskerin, mutated in X-linked DC, in human ribosome biogenesis and p53 regulation, and to explore the link between ribosome production and p53 homeostasis. Dyskerin is an rRNA pseudouridine synthase and required for telomere maintenance. There is some debate as to whether DC is the result of telomere maintenance or ribosome biogenesis defects. It is shown here that human Dyskerin is required for the production of both LSU and SSU, and knockdown of Dyskerin leads to p53 activation via inhibition of MDM2 via the 5S RNP, an LSU assembly intermediate which accumulates after ribosome biogenesis defects. My data indicate that p53 activation, due to defects in ribosome biogenesis, may contribute to the clinical symptoms seen in patients suffering with DC. In addition, it is shown that defects in early or late stages of SSU or LSU biogenesis, result in activation of p53 via the 5S RNP-MDM2 pathway, and that p53 is induced in less than 12 hours after ribosome biogenesis defects. -

Colony PCR Amplification of the 16S Ribosomal RNA Gene I. OBJECTIVES
Lab: Colony PCR amplification of the 16S ribosomal RNA gene I. OBJECTIVES (1) Learn how to use the polymerase chain reaction (PCR) to amplify the small subunit ribosomal RNA (SSU rRNA) gene from a bacterial colony; (2) Learn how to run an agarose gel to visualize the resulting PCR amplicons and extract the amplified DNA from the agarose gel. II. INTRODUCTION Microbial community structure is a critical determinant of the biogeochemical processes occurring within the pelagic marine ecosystem. Due to the difficulties of cultivating many marine microbes, microbial ecologists frequently employ molecular biology-based techniques to reveal the complexity of aquatic microbial communities. Culture-independent techniques for characterizing microbial biodiversity are primarily based on the analysis of small subunit ribosomal RNA (SSU rRNA) genes from environmental samples (e.g. Giovannoni et al. 1990). This gene is an excellent phylogenetic marker for Bacteria and Archaea. With the aid of the polymerase chain reaction (PCR), these strategies have greatly enhanced our ability to describe the genetic diversity of microorganisms in the natural environment without the need for cultivation. The cloning and sequencing of PCR-amplified SSU rRNA genes is now a routine procedure. An overview of the typical steps is as follows: 1) SSU rDNAs (rDNA = rRNA gene) are amplified via the polymerase chain reaction using conserved oligonucleotide primers, 2) amplified products are cloned into a plasmid vector, 3) E. coli cell are transformed with the recombinant vectors 4) individual colonies of transformed E. coli are picked and the plasmids are purified 5) the purified plasmids are used as a template for the sequencing reaction. -

LETM1 Haploinsufficiency Causes Mitochondrial Defects in Cells From
© 2014. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2014) 7, 535-545 doi:10.1242/dmm.014464 RESEARCH ARTICLE LETM1 haploinsufficiency causes mitochondrial defects in cells from humans with Wolf-Hirschhorn syndrome: implications for dissecting the underlying pathomechanisms in this condition Lesley Hart1,2, Anita Rauch3, Antony M. Carr2, Joris R. Vermeesch4 and Mark O’Driscoll1,* ABSTRACT abnormalities, a characteristic facial dysmorphology, hypotonia, and Wolf-Hirschhorn syndrome (WHS) represents an archetypical epileptic seizures (Hirschhorn and Cooper, 1961; Hirschhorn et al., example of a contiguous gene deletion disorder – a condition 1965; Wolf et al., 1965). The spectrum and severity of these clinical comprising a complex set of developmental phenotypes with a features typically correlate with deletion size (Battaglia et al., 2008; multigenic origin. Epileptic seizures, intellectual disability, growth Maas et al., 2008; Van Buggenhout et al., 2004; Zollino et al., 2000). restriction, motor delay and hypotonia are major co-morbidities in WHS is generally regarded as a multigenic disorder, although two WHS. Haploinsufficiency of LETM1, which encodes a mitochondrial critical regions have been described: WHSCR1 and WHSCR2, for inner-membrane protein functioning in ion transport, has been WHS critical region 1 and 2, respectively (see Fig. 1). These critical proposed as an underlying pathomechanism, principally for seizures regions are based on the demarcation of the minimum region of but also for other core features of WHS, including growth and motor overlap in individuals exhibiting WHS-like phenotypes. WHSCR1 delay. Growing evidence derived from several model organisms incorporates part of the WHS candidate gene WHSC1 and the entire suggests that reduced LETM1 expression is associated with some WHSC2 gene (White et al., 1995; Wright et al., 1997). -

Ribosomal DNA Copy Loss and Repeat Instability in ATRX-Mutated Cancers
Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers Maheshi Udugamaa,1, Elaine Sanijb,c,1, Hsiao P. J. Voona, Jinbae Sonb, Linda Hiia, Jeremy D. Hensond, F. Lyn Chana, Fiona T. M. Changa, Yumei Liue, Richard B. Pearsona,b,f,g, Paul Kalitsish, Jeffrey R. Manni, Philippe Collasj,k, Ross D. Hannana,b,f,g,l, and Lee H. Wonga,2 aDepartment of Biochemistry and Molecular Biology, Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia; bResearch Division, Peter MacCallum Cancer Centre, Parkville, VIC 2010, Australia; cDepartment of Pathology, The University of Melbourne, Parkville, VIC 3010, Australia; dCancer Cell Immortality Group, Adult Cancer Program, Prince of Wales Clinical School, University of New South Wales, Randwick, NSW 2052, Australia; eCollege of Animal Science and Technology, Henan University of Science and Technology, Luoyang, Henan Province, 471023, China; fSir Peter MacCallum Department of Oncology, The University of Melbourne, VIC 3010, Australia; gDepartment of Biochemistry and Molecular Biology, The University of Melbourne, Parkville, VIC 3010, Australia; hDepartment of Paediatrics, Murdoch Children’s Research Institute, Royal Children’s Hospital, University of Melbourne, Parkville, VIC 3052, Australia; iGenome Modification Platform, Monash University, Clayton, VIC 3800, Australia; jDepartment of Molecular Medicine, Institute of Basic Medical Sciences, Faculty of Medicine, University of Oslo, 0317 Oslo, Norway; kNorwegian Center for Stem Cell Research, Department of Immunology