Diverse Entry Gates to Mitochondria Provide Insights Into the Evolution of Eukaryotes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rise and Fall of the Bovine Corpus Luteum
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Rise and Fall of the Bovine Corpus Luteum Heather Talbott University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, Molecular Biology Commons, and the Obstetrics and Gynecology Commons Recommended Citation Talbott, Heather, "The Rise and Fall of the Bovine Corpus Luteum" (2017). Theses & Dissertations. 207. https://digitalcommons.unmc.edu/etd/207 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE RISE AND FALL OF THE BOVINE CORPUS LUTEUM by Heather Talbott A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor John S. Davis University of Nebraska Medical Center Omaha, Nebraska May, 2017 Supervisory Committee: Carol A. Casey, Ph.D. Andrea S. Cupp, Ph.D. Parmender P. Mehta, Ph.D. Justin L. Mott, Ph.D. i ACKNOWLEDGEMENTS This dissertation was supported by the Agriculture and Food Research Initiative from the USDA National Institute of Food and Agriculture (NIFA) Pre-doctoral award; University of Nebraska Medical Center Graduate Student Assistantship; University of Nebraska Medical Center Exceptional Incoming Graduate Student Award; the VA Nebraska-Western Iowa Health Care System Department of Veterans Affairs; and The Olson Center for Women’s Health, Department of Obstetrics and Gynecology, Nebraska Medical Center. -

What Are Their Roles in Mitochondrial Protein Synthesis?
Characterisation of human mtRF1 and C12orf65: What are their roles in mitochondrial protein synthesis? Aleksandra Pajak M.Res Thesis submitted to Newcastle University in candidature for the degree of Doctor of Philosophy Newcastle University Faculty of Medical Sciences Institute for Ageing and Health Mitochondrial Research Group January 2013 Abstract Mitochondria have their own protein synthesis machinery that synthesises the oxidative phosphorylation components encoded by their mtDNA. This translation process consists of four main phases: initiation, elongation, termination and ribosome recycling. Termination and its control have been the least investigated. Recently, however, the termination factor, mtRF1a, has been characterised as sufficient to release all the nascent proteins from the mitoribosome. Furthermore, bioinformatics has identified three additional members of this mitochondrial release factor family namely, mtRF1, C12orf65 and ICT1. The latter is now known to be incorporated into the mitoribosome but its exact function remains unclear. My project has therefore focussed on characterising the remaining two factors; mtRF1 and C12orf65, and investigating their possible involvement in mitochondrial protein synthesis. It has been demonstrated that protein synthesis is not perfect and bacterial ribosomes not infrequently stall during translation. This can result from limiting amounts of charged tRNAs, stable secondary structures, or truncated/degraded transcripts. Ribosome stalling has been shown to cause growth arrest. In order to prevent that and maintain high efficiency of mitochondrial protein synthesis such stalled complexes need to be rapidly recycled. Bacteria have developed at least three distinct mechanisms by which ribosomes can be rescued. Contrastingly, despite the presence of truncated mRNAs in mitochondria, no such quality control mechanisms have been identified in these organelles. -

Dual Role of the Mitochondrial Protein Frataxin in Astrocytic Tumors
Laboratory Investigation (2011) 91, 1766–1776 & 2011 USCAP, Inc All rights reserved 0023-6837/11 $32.00 Dual role of the mitochondrial protein frataxin in astrocytic tumors Elmar Kirches1, Nadine Andrae1, Aline Hoefer2, Barbara Kehler1,3, Kim Zarse4, Martin Leverkus3, Gerburg Keilhoff5, Peter Schonfeld5, Thomas Schneider6, Annette Wilisch-Neumann1 and Christian Mawrin1 The mitochondrial protein frataxin (FXN) is known to be involved in mitochondrial iron homeostasis and iron–sulfur cluster biogenesis. It is discussed to modulate function of the electron transport chain and production of reactive oxygen species (ROS). FXN loss in neurons and heart muscle cells causes an autosomal-dominant mitochondrial disorder, Friedreich’s ataxia. Recently, tumor induction after targeted FXN deletion in liver and reversal of the tumorigenic phenotype of colonic carcinoma cells following FXN overexpression were described in the literature, suggesting a tumor suppressor function. We hypothesized that a partial reversal of the malignant phenotype of glioma cells should occur after FXN transfection, if the mitochondrial protein has tumor suppressor functions in these brain tumors. In astrocytic brain tumors and tumor cell lines, we observed reduced FXN levels compared with non-neoplastic astrocytes. Mitochondrial content (citrate synthase activity) was not significantly altered in U87MG glioblastoma cells stably overexpressing FXN (U87-FXN). Surprisingly, U87-FXN cells exhibited increased cytoplasmic ROS levels, although mitochondrial ROS release was attenuated by FXN, as expected. Higher cytoplasmic ROS levels corresponded to reduced activities of glutathione peroxidase and catalase, and lower glutathione content. The defect of antioxidative capacity resulted in increased susceptibility of U87-FXN cells against oxidative stress induced by H2O2 or buthionine sulfoximine. -

Evaluation of Cancer-Derived Myocardial Impairments Using a Mouse Model
www.oncotarget.com Oncotarget, 2020, Vol. 11, (No. 41), pp: 3712-3722 Research Paper Evaluation of cancer-derived myocardial impairments using a mouse model Yoshihiro Miyagawa1, Shota Nukaga1,2, Takuya Mori1, Rina Fujiwara-Tani1, Kiyomu Fujii1, Shiori Mori1, Kei Goto1,3, Shingo Kishi1, Takamitsu Sasaki1, Chie Nakashima1, Hitoshi Ohmori1, Isao Kawahara1,2, Yi Luo4 and Hiroki Kuniyasu1 1Department of Molecular Pathology, Nara Medical University, Kashihara, Nara 634-8521, Japan 2Division of Rehabilitation, Hanna Central Hospital, Ikoma, Nara 630-0243, Japan 3Division of Rehabilitation, Hoshida Minami Hospital, Katano, Osaka 576-0022, Japan 4Key Laboratory of Neuroregeneration of Jiangsu and Ministry of Education, Co-Innovation Center of Neuroregeneration, Nantong University, Nantong, Jiangsu Province 226001, China Correspondence to: Yi Luo, email: [email protected] Hiroki Kuniyasu, email: [email protected] Keywords: cachexia; myocardium; atrophy; mitochondria; oxidative stress Received: June 26, 2020 Accepted: September 10, 2020 Published: October 13, 2020 Copyright: © 2020 Miyagawa et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Myocardial damage in cancer patients is emphasized as a cause of death; however, there are not many murine cachexia models to evaluate cancer-derived heart disorder. Using the mouse cachexia model that we established previously, we investigated myocardial damage in tumor-bearing mice. In cachexic mice, decreased heart weight and myocardial volume, and dilated left ventricular lumen, and atrophied cardiomyocytes were noted. The cardiomyocytes also showed accumulated 8-hydroxydeoxyguanosine, decreased leucine zipper and EF-hand- containing transmembrane protein-1, and increased microtubule-associated protein light chain3-II. -

The Pathogenetic Role of Β-Cell Mitochondria in Type 2 Diabetes
236 3 Journal of M Fex et al. Mitochondria in β-cells 236:3 R145–R159 Endocrinology REVIEW The pathogenetic role of β-cell mitochondria in type 2 diabetes Malin Fex1, Lisa M Nicholas1, Neelanjan Vishnu1, Anya Medina1, Vladimir V Sharoyko1, David G Nicholls1, Peter Spégel1,2 and Hindrik Mulder1 1Department of Clinical Sciences in Malmö, Unit of Molecular Metabolism, Lund University Diabetes Centre, Clinical Research Center, Malmö University Hospital, Lund University, Malmö, Sweden 2Department of Chemistry, Center for Analysis and Synthesis, Lund University, Sweden Correspondence should be addressed to H Mulder: [email protected] Abstract Mitochondrial metabolism is a major determinant of insulin secretion from pancreatic Key Words β-cells. Type 2 diabetes evolves when β-cells fail to release appropriate amounts f TCA cycle of insulin in response to glucose. This results in hyperglycemia and metabolic f coupling signal dysregulation. Evidence has recently been mounting that mitochondrial dysfunction f oxidative phosphorylation plays an important role in these processes. Monogenic dysfunction of mitochondria is a f mitochondrial transcription rare condition but causes a type 2 diabetes-like syndrome owing to β-cell failure. Here, f genetic variation we describe novel advances in research on mitochondrial dysfunction in the β-cell in type 2 diabetes, with a focus on human studies. Relevant studies in animal and cell models of the disease are described. Transcriptional and translational regulation in mitochondria are particularly emphasized. The role of metabolic enzymes and pathways and their impact on β-cell function in type 2 diabetes pathophysiology are discussed. The role of genetic variation in mitochondrial function leading to type 2 diabetes is highlighted. -

LETM1 Gene Leucine Zipper and EF-Hand Containing Transmembrane Protein 1
LETM1 gene leucine zipper and EF-hand containing transmembrane protein 1 Normal Function The LETM1 gene provides instructions for making a protein whose function is not well understood. This protein is active in mitochondria, which are structures within cells that convert the energy from food into a form that cells can use. The LETM1 protein may be involved in the transport of charged calcium atoms (calcium ions) across membranes within mitochondria. Researchers suspect that the protein also plays a role in determining the shape and volume of mitochondria. Health Conditions Related to Genetic Changes Wolf-Hirschhorn syndrome The LETM1 gene is located in a region of chromosome 4 that is deleted in people with the typical features of Wolf-Hirschhorn syndrome. As a result of this deletion, affected individuals are missing one copy of the LETM1 gene in each cell. Studies suggest that a loss of this gene alters the structure of mitochondria; however, it is unclear how this abnormality is related to the signs and symptoms of Wolf-Hirschhorn syndrome. Specifically, a loss of the LETM1 gene has been associated with seizures or other abnormal electrical activity in the brain. Other Names for This Gene • LETM1_HUMAN • leucine zipper-EF-hand containing transmembrane protein 1 Additional Information & Resources Tests Listed in the Genetic Testing Registry • Tests of LETM1 (https://www.ncbi.nlm.nih.gov/gtr/all/tests/?term=3954[geneid]) Scientific Articles on PubMed Reprinted from MedlinePlus Genetics (https://medlineplus.gov/genetics/) 1 • PubMed (https://pubmed.ncbi.nlm.nih.gov/?term=%28%28LETM1%5BTIAB%5D%2 -

Src Inhibitors Modulate Frataxin Protein Levels
Human Molecular Genetics, 2015, Vol. 24, No. 15 4296–4305 doi: 10.1093/hmg/ddv162 Advance Access Publication Date: 6 May 2015 Original Article ORIGINAL ARTICLE Src inhibitors modulate frataxin protein levels Downloaded from https://academic.oup.com/hmg/article/24/15/4296/2453025 by guest on 28 September 2021 Fabio Cherubini1, Dario Serio1, Ilaria Guccini1, Silvia Fortuni1,2, Gaetano Arcuri1, Ivano Condò1, Alessandra Rufini1,2, Shadman Moiz1, Serena Camerini3, Marco Crescenzi3, Roberto Testi1,2 and Florence Malisan1,* 1Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy, 2Fratagene Therapeutics Ltd, 22 Northumberland Rd, Dublin, Ireland and 3Department of Cell Biology and Neurosciences, Italian National Institute of Health, Viale Regina Elena, 299, 00161 Rome, Italy *To whom correspondence should be addressed at: Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy. Tel: +39 0672596501; Fax: +39 0672596505; Email: [email protected] Abstract Defective expression of frataxin is responsible for the inherited, progressive degenerative disease Friedreich’s Ataxia (FRDA). There is currently no effective approved treatment for FRDA and patients die prematurely. Defective frataxin expression causes critical metabolic changes, including redox imbalance and ATP deficiency. As these alterations are known to regulate the tyrosine kinase Src, we investigated whether Src might in turn affect frataxin expression. We found that frataxin can be phosphorylated by Src. Phosphorylation occurs primarily on Y118 and promotes frataxin ubiquitination, a signal for degradation. Accordingly, Src inhibitors induce accumulation of frataxin but are ineffective on a non-phosphorylatable frataxin- Y118F mutant. -
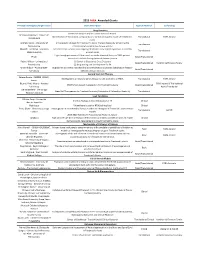
Grantsummaries 2014-2017 Jf
2015 FARA Awarded Grants Principal Investigator/Organization Grant Description Type of Research Co-funding Drug Discovery 2014 Keith Michael Andrus Cardiac Research Award: Veronique Monnier - Université Identification of therapeutic compounds on a cardiac Drosophila model of Friedreich’s Translational FARA Ireland Paris Diderot ataxia Andrew Dancis - University of A therapeutic strategy for Friedreich's ataxia: Frataxin bypass by enhancing the Translational Pennsylvania mitochondrial cysteine desulfurase activity Edward L. Grabczyk - Louisiana Small molecule induced exon skipping of MLH3 to slow repeat expansion in an FRDA Translational State University mousel model High throughput screen of Pfizer small molecule chemical library in FRDA patient- Pfizer Basic/Translational derived cells to look for regulators of frataxin protein Robert Wilson - University of (1) Center of Excellence: Drug Discovery Basic/Translational Hamilton & Finneran Family Pennsylvania (2) Drug testing and development for FA Omar Khdour - Arizona State Approaches to correct mitochondrial abnormalities and synaptic pathology in frataxin- Basic/Translational University deficient sensory neurons Gene & Stem Cell Therapy Helene Puccio - INSERM, IGBMC, Development of neuronal gene therapy for the treatment of FRDA Translational FARA Ireland France Ricardo Pinto Mouro - Harvard FARA Ireland & The National CRISPR/Cas9-based Therapeutics for Friedreich's ataxia Basic/Translational University Ataxia Foundation Joel Gottesfeld - The Scripps Stem Cell Therapeutics for Friedreich's -

Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’S Disease
biomedicines Review Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease Heng-Chung Kung 1,2 , Kai-Jung Lin 1,3 , Chia-Te Kung 4,* and Tsu-Kung Lin 1,5,6,* 1 Center for Mitochondrial Research and Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan; [email protected] (H.-C.K.); [email protected] (K.-J.L.) 2 Department of Biology, Krieger School of Arts and Sciences, Johns Hopkins University, Baltimore, MD 21218, USA 3 Department of Family Medicine, National Taiwan University Hospital, Taipei 100225, Taiwan 4 Department of Emergency Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan 5 Department of Neurology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan 6 Center of Parkinson’s Disease, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan * Correspondence: [email protected] (C.-T.K.); [email protected] (T.-K.L.) Abstract: Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by dopaminergic neuronal loss. The exact pathogenesis of PD is complex and not yet completely understood, but research has established the critical role mitochondrial dysfunction plays in the development of PD. As the main producer of cytosolic reactive oxygen species (ROS), mito- chondria are particularly susceptible to oxidative stress once an imbalance between ROS generation and the organelle’s antioxidative system occurs. An overabundance of ROS in the mitochondria can Citation: Kung, H.-C.; Lin, K.-J.; lead to mitochondrial dysfunction and further vicious cycles. -

LETM1 Haploinsufficiency Causes Mitochondrial Defects in Cells From
© 2014. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2014) 7, 535-545 doi:10.1242/dmm.014464 RESEARCH ARTICLE LETM1 haploinsufficiency causes mitochondrial defects in cells from humans with Wolf-Hirschhorn syndrome: implications for dissecting the underlying pathomechanisms in this condition Lesley Hart1,2, Anita Rauch3, Antony M. Carr2, Joris R. Vermeesch4 and Mark O’Driscoll1,* ABSTRACT abnormalities, a characteristic facial dysmorphology, hypotonia, and Wolf-Hirschhorn syndrome (WHS) represents an archetypical epileptic seizures (Hirschhorn and Cooper, 1961; Hirschhorn et al., example of a contiguous gene deletion disorder – a condition 1965; Wolf et al., 1965). The spectrum and severity of these clinical comprising a complex set of developmental phenotypes with a features typically correlate with deletion size (Battaglia et al., 2008; multigenic origin. Epileptic seizures, intellectual disability, growth Maas et al., 2008; Van Buggenhout et al., 2004; Zollino et al., 2000). restriction, motor delay and hypotonia are major co-morbidities in WHS is generally regarded as a multigenic disorder, although two WHS. Haploinsufficiency of LETM1, which encodes a mitochondrial critical regions have been described: WHSCR1 and WHSCR2, for inner-membrane protein functioning in ion transport, has been WHS critical region 1 and 2, respectively (see Fig. 1). These critical proposed as an underlying pathomechanism, principally for seizures regions are based on the demarcation of the minimum region of but also for other core features of WHS, including growth and motor overlap in individuals exhibiting WHS-like phenotypes. WHSCR1 delay. Growing evidence derived from several model organisms incorporates part of the WHS candidate gene WHSC1 and the entire suggests that reduced LETM1 expression is associated with some WHSC2 gene (White et al., 1995; Wright et al., 1997). -

Strand Breaks for P53 Exon 6 and 8 Among Different Time Course of Folate Depletion Or Repletion in the Rectosigmoid Mucosa
SUPPLEMENTAL FIGURE COLON p53 EXONIC STRAND BREAKS DURING FOLATE DEPLETION-REPLETION INTERVENTION Supplemental Figure Legend Strand breaks for p53 exon 6 and 8 among different time course of folate depletion or repletion in the rectosigmoid mucosa. The input of DNA was controlled by GAPDH. The data is shown as ΔCt after normalized to GAPDH. The higher ΔCt the more strand breaks. The P value is shown in the figure. SUPPLEMENT S1 Genes that were significantly UPREGULATED after folate intervention (by unadjusted paired t-test), list is sorted by P value Gene Symbol Nucleotide P VALUE Description OLFM4 NM_006418 0.0000 Homo sapiens differentially expressed in hematopoietic lineages (GW112) mRNA. FMR1NB NM_152578 0.0000 Homo sapiens hypothetical protein FLJ25736 (FLJ25736) mRNA. IFI6 NM_002038 0.0001 Homo sapiens interferon alpha-inducible protein (clone IFI-6-16) (G1P3) transcript variant 1 mRNA. Homo sapiens UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 15 GALNTL5 NM_145292 0.0001 (GALNT15) mRNA. STIM2 NM_020860 0.0001 Homo sapiens stromal interaction molecule 2 (STIM2) mRNA. ZNF645 NM_152577 0.0002 Homo sapiens hypothetical protein FLJ25735 (FLJ25735) mRNA. ATP12A NM_001676 0.0002 Homo sapiens ATPase H+/K+ transporting nongastric alpha polypeptide (ATP12A) mRNA. U1SNRNPBP NM_007020 0.0003 Homo sapiens U1-snRNP binding protein homolog (U1SNRNPBP) transcript variant 1 mRNA. RNF125 NM_017831 0.0004 Homo sapiens ring finger protein 125 (RNF125) mRNA. FMNL1 NM_005892 0.0004 Homo sapiens formin-like (FMNL) mRNA. ISG15 NM_005101 0.0005 Homo sapiens interferon alpha-inducible protein (clone IFI-15K) (G1P2) mRNA. SLC6A14 NM_007231 0.0005 Homo sapiens solute carrier family 6 (neurotransmitter transporter) member 14 (SLC6A14) mRNA. -

Stríbrná and Julius Lukes Hassan Hashimi, Lindsay Mcdonald, Eva
Bioenergetics: Trypanosome Letm1 Protein Is Essential for Mitochondrial Potassium Homeostasis Hassan Hashimi, Lindsay McDonald, Eva Stríbrná and Julius Lukes J. Biol. Chem. 2013, 288:26914-26925. doi: 10.1074/jbc.M113.495119 originally published online July 26, 2013 Access the most updated version of this article at doi: 10.1074/jbc.M113.495119 Find articles, minireviews, Reflections and Classics on similar topics on the JBC Affinity Sites. Alerts: • When this article is cited • When a correction for this article is posted Click here to choose from all of JBC's e-mail alerts Supplemental material: http://www.jbc.org/content/suppl/2013/07/26/M113.495119.DC1.html This article cites 60 references, 26 of which can be accessed free at http://www.jbc.org/content/288/37/26914.full.html#ref-list-1 Downloaded from http://www.jbc.org/ at Edinburgh University Library on September 13, 2013 THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 288, NO. 37, pp. 26914–26925, September 13, 2013 © 2013 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A. Trypanosome Letm1 Protein Is Essential for Mitochondrial Potassium Homeostasis*□S Received for publication, June 19, 2013, and in revised form, July 23, 2013 Published, JBC Papers in Press, July 26, 2013, DOI 10.1074/jbc.M113.495119 Hassan Hashimi‡§1, Lindsay McDonald‡2, Eva Strˇíbrná‡, and Julius Lukesˇ‡§3 From the ‡Institute of Parasitology, Biology Centre, Czech Academy of Sciences and the §Faculty of Science, University of South Bohemia, 370 05 Cˇeské Budeˇjovice (Budweis), Czech Republic Background: Letm1 is a mitochondrial protein attributed disparate roles, including cation/proton antiport and translation.