The Pathogenetic Role of Β-Cell Mitochondria in Type 2 Diabetes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Altered Expression and Function of Mitochondrial Я-Oxidation Enzymes
0031-3998/01/5001-0083 PEDIATRIC RESEARCH Vol. 50, No. 1, 2001 Copyright © 2001 International Pediatric Research Foundation, Inc. Printed in U.S.A. Altered Expression and Function of Mitochondrial -Oxidation Enzymes in Juvenile Intrauterine-Growth-Retarded Rat Skeletal Muscle ROBERT H. LANE, DAVID E. KELLEY, VLADIMIR H. RITOV, ANNA E. TSIRKA, AND ELISA M. GRUETZMACHER Department of Pediatrics, UCLA School of Medicine, Mattel Children’s Hospital at UCLA, Los Angeles, California 90095, U.S.A. [R.H.L.]; and Departments of Internal Medicine [D.E.K., V.H.R.] and Pediatrics [R.H.L., A.E.T., E.M.G.], University of Pittsburgh School of Medicine, Magee-Womens Research Institute, Pittsburgh, Pennsylvania 15213, U.S.A. ABSTRACT Uteroplacental insufficiency and subsequent intrauterine creased in IUGR skeletal muscle mitochondria, and isocitrate growth retardation (IUGR) affects postnatal metabolism. In ju- dehydrogenase activity was unchanged. Interestingly, skeletal venile rats, IUGR alters skeletal muscle mitochondrial gene muscle triglycerides were significantly increased in IUGR skel- expression and reduces mitochondrial NADϩ/NADH ratios, both etal muscle. We conclude that uteroplacental insufficiency alters of which affect -oxidation flux. We therefore hypothesized that IUGR skeletal muscle mitochondrial lipid metabolism, and we gene expression and function of mitochondrial -oxidation en- speculate that the changes observed in this study play a role in zymes would be altered in juvenile IUGR skeletal muscle. To test the long-term morbidity associated with IUGR. (Pediatr Res 50: this hypothesis, mRNA levels of five key mitochondrial enzymes 83–90, 2001) (carnitine palmitoyltransferase I, trifunctional protein of -oxi- dation, uncoupling protein-3, isocitrate dehydrogenase, and mi- Abbreviations tochondrial malate dehydrogenase) and intramuscular triglycer- CPTI, carnitine palmitoyltransferase I ides were quantified in 21-d-old (preweaning) IUGR and control IUGR, intrauterine growth retardation rat skeletal muscle. -

Mt-Atp8 Gene in the Conplastic Mouse Strain C57BL/6J-Mtfvb/NJ on the Mitochondrial Function and Consequent Alterations to Metabolic and Immunological Phenotypes
From the Lübeck Institute of Experimental Dermatology of the University of Lübeck Director: Prof. Dr. Saleh M. Ibrahim Interplay of mtDNA, metabolism and microbiota in the pathogenesis of AIBD Dissertation for Fulfillment of Requirements for the Doctoral Degree of the University of Lübeck from the Department of Natural Sciences Submitted by Paul Schilf from Rostock Lübeck, 2016 First referee: Prof. Dr. Saleh M. Ibrahim Second referee: Prof. Dr. Stephan Anemüller Chairman: Prof. Dr. Rainer Duden Date of oral examination: 30.03.2017 Approved for printing: Lübeck, 06.04.2017 Ich versichere, dass ich die Dissertation ohne fremde Hilfe angefertigt und keine anderen als die angegebenen Hilfsmittel verwendet habe. Weder vorher noch gleichzeitig habe ich andernorts einen Zulassungsantrag gestellt oder diese Dissertation vorgelegt. ABSTRACT Mitochondria are critical in the regulation of cellular metabolism and influence signaling processes and inflammatory responses. Mitochondrial DNA mutations and mitochondrial dysfunction are known to cause a wide range of pathological conditions and are associated with various immune diseases. The findings in this work describe the effect of a mutation in the mitochondrially encoded mt-Atp8 gene in the conplastic mouse strain C57BL/6J-mtFVB/NJ on the mitochondrial function and consequent alterations to metabolic and immunological phenotypes. This work provides insights into the mutation-induced cellular adaptations that influence the inflammatory milieu and shape pathological processes, in particular focusing on autoimmune bullous diseases, which have recently been reported to be associated with mtDNA polymorphisms in the human MT-ATP8 gene. The mt-Atp8 mutation diminishes the assembly of the ATP synthase complex into multimers and decreases mitochondrial respiration, affects generation of reactive oxygen species thus leading to a shift in the metabolic balance and reduction in the energy state of the cell as indicated by the ratio ATP to ADP. -

The Emerging Neuroprotective Role of Mitochondrial Uncoupling Protein
Review Article • DOI: 10.1515/tnsci-2015-0019 • Translational Neuroscience • 6 • 2015 • 179-186 Translational Neuroscience THE EMERGING NEUROPROTECTIVE ROLE Kieran P. Normoyle1,2,3, OF MITOCHONDRIAL Miri Kim2,4, Arash Farahvar5, UNCOUPLING PROTEIN-2 Daniel Llano1,6,7, Kevin Jackson7,8, IN TRAUMATIC BRAIN INJURY Huan Wang6,8* Abstract 1Department of Molecular and Integrative Traumatic brain injury (TBI) is a multifaceted disease with intrinsically complex heterogeneity and remains a Physiology, University of Illinois at Urbana- Champaign, Urbana, IL, USA significant clinical challenge to manage. TBI model systems have demonstrated many mechanisms that contribute 2College of Medicine, University of Illinois at to brain parenchymal cell death, including glutamate and calcium toxicity, oxidative stress, inflammation, and Urbana-Champaign, Urbana, IL, USA mitochondrial dysfunction. Mitochondria are critically regulated by uncoupling proteins (UCP), which allow 3Department of Child Neurology, Massachusetts protons to leak back into the matrix and thus reduce the mitochondrial membrane potential by dissipating the General Hospital, Boston, MA, USA 4Department of Cell and Developmental Biology, proton motive force. This uncoupling of oxidative phosphorylation from adenosine triphosphate (ATP) synthesis University of Illinois at Urbana-Champaign, is potentially critical for protection against cellular injury as a result of TBI and stroke. A greater understanding Urbana, IL, USA of the underlying mechanism or mechanisms by which uncoupling protein-2 -

Dual Role of the Mitochondrial Protein Frataxin in Astrocytic Tumors
Laboratory Investigation (2011) 91, 1766–1776 & 2011 USCAP, Inc All rights reserved 0023-6837/11 $32.00 Dual role of the mitochondrial protein frataxin in astrocytic tumors Elmar Kirches1, Nadine Andrae1, Aline Hoefer2, Barbara Kehler1,3, Kim Zarse4, Martin Leverkus3, Gerburg Keilhoff5, Peter Schonfeld5, Thomas Schneider6, Annette Wilisch-Neumann1 and Christian Mawrin1 The mitochondrial protein frataxin (FXN) is known to be involved in mitochondrial iron homeostasis and iron–sulfur cluster biogenesis. It is discussed to modulate function of the electron transport chain and production of reactive oxygen species (ROS). FXN loss in neurons and heart muscle cells causes an autosomal-dominant mitochondrial disorder, Friedreich’s ataxia. Recently, tumor induction after targeted FXN deletion in liver and reversal of the tumorigenic phenotype of colonic carcinoma cells following FXN overexpression were described in the literature, suggesting a tumor suppressor function. We hypothesized that a partial reversal of the malignant phenotype of glioma cells should occur after FXN transfection, if the mitochondrial protein has tumor suppressor functions in these brain tumors. In astrocytic brain tumors and tumor cell lines, we observed reduced FXN levels compared with non-neoplastic astrocytes. Mitochondrial content (citrate synthase activity) was not significantly altered in U87MG glioblastoma cells stably overexpressing FXN (U87-FXN). Surprisingly, U87-FXN cells exhibited increased cytoplasmic ROS levels, although mitochondrial ROS release was attenuated by FXN, as expected. Higher cytoplasmic ROS levels corresponded to reduced activities of glutathione peroxidase and catalase, and lower glutathione content. The defect of antioxidative capacity resulted in increased susceptibility of U87-FXN cells against oxidative stress induced by H2O2 or buthionine sulfoximine. -

The Role of Uncoupling Protein 3 in Human Physiology
The role of uncoupling protein 3 in human physiology W. Timothy Garvey J Clin Invest. 2003;111(4):438-441. https://doi.org/10.1172/JCI17835. Commentary Obesity is simply understood as an imbalance between energy intake and expenditure in favor of weight accretion. However, the human biological interface between food consumption and energy dissipation results in broad individual differences in eating behavior, physical activity, and efficiency of fuel storage and metabolism. In particular, the basal metabolic rate, which accounts for the greatest portion of overall energy expenditure, can vary almost twofold among individuals. Classically, three major biochemical systems are believed to contribute to basal thermogenesis: futile cycles, Na+/K+ATPase activity, and mitochondrial proton leak. The latter is the most important quantitative contributor and can explain up to 50% of the basal metabolic rate (1). The molecular basis of mitochondrial proton leak is unclear, despite its importance in the understanding of energy balance and its potential as a therapeutic target for obesity treatment. The article by Hesselink and colleagues in this issue of the JCI (2) addresses whether uncoupling protein 3 contributes to mitochondrial proton leak in human skeletal muscle. Mitochondrial respiration and oxidative phosphorylation The oxidation of fatty acids and pyruvate takes place in mitochondria, where energy is converted into ATP for use in cellular processes. Reducing equivalents are extracted from substrates and sequentially passed from electron donors (reductants) to acceptors (oxidants) along the mitochondrial respiratory chain to molecular oxygen. The electron transport system is located on […] Find the latest version: https://jci.me/17835/pdf COMMENTARY See the related article beginning on page 479. -

SARS-Cov-2 Restructures the Host Chromatin Architecture 2
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.20.453146; this version posted July 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 SARS-CoV-2 Restructures the Host Chromatin Architecture 2 3 Authors: Ruoyu Wang 1,2,†, Joo-Hyung Lee 1,†, Feng Xiong 1, Jieun Kim 3,4, Lana Al Hasani 1,2, Xiaoyi 4 Yuan 3,4, Pooja Shivshankar 1,3,4, Joanna Krakowiak 1, Chuangye Qi 1, Yanyu Wang 3,4, Holger K. 5 Eltzschig 2,3,4, Wenbo Li 1,2,* 6 7 Affiliations: 8 1 Department of Biochemistry and Molecular Biology, McGovern Medical School, University of Texas 9 Health Science Center, Houston, 77030, TX, USA 10 2 Graduate School of Biomedical Sciences, University of Texas MD Anderson Cancer Center and 11 UTHealth, Houston, 77030, TX, USA 12 3 Department of Anesthesiology, McGovern Medical School, University of Texas Health Science Center, 13 Houston, 77030, TX, USA 14 4 Center for Perioperative Medicine, McGovern Medical School, University of Texas Health Science 15 Center, Houston, 77030, TX, USA 16 17 † These authors contributed equally 18 * Correspondence: Wenbo Li, Ph.D. ([email protected]). MSB 6.161, Fannin Street, Houston, 19 Texas, 77030, USA; Tel: 713-500-6103. 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.07.20.453146; this version posted July 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Src Inhibitors Modulate Frataxin Protein Levels
Human Molecular Genetics, 2015, Vol. 24, No. 15 4296–4305 doi: 10.1093/hmg/ddv162 Advance Access Publication Date: 6 May 2015 Original Article ORIGINAL ARTICLE Src inhibitors modulate frataxin protein levels Downloaded from https://academic.oup.com/hmg/article/24/15/4296/2453025 by guest on 28 September 2021 Fabio Cherubini1, Dario Serio1, Ilaria Guccini1, Silvia Fortuni1,2, Gaetano Arcuri1, Ivano Condò1, Alessandra Rufini1,2, Shadman Moiz1, Serena Camerini3, Marco Crescenzi3, Roberto Testi1,2 and Florence Malisan1,* 1Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy, 2Fratagene Therapeutics Ltd, 22 Northumberland Rd, Dublin, Ireland and 3Department of Cell Biology and Neurosciences, Italian National Institute of Health, Viale Regina Elena, 299, 00161 Rome, Italy *To whom correspondence should be addressed at: Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy. Tel: +39 0672596501; Fax: +39 0672596505; Email: [email protected] Abstract Defective expression of frataxin is responsible for the inherited, progressive degenerative disease Friedreich’s Ataxia (FRDA). There is currently no effective approved treatment for FRDA and patients die prematurely. Defective frataxin expression causes critical metabolic changes, including redox imbalance and ATP deficiency. As these alterations are known to regulate the tyrosine kinase Src, we investigated whether Src might in turn affect frataxin expression. We found that frataxin can be phosphorylated by Src. Phosphorylation occurs primarily on Y118 and promotes frataxin ubiquitination, a signal for degradation. Accordingly, Src inhibitors induce accumulation of frataxin but are ineffective on a non-phosphorylatable frataxin- Y118F mutant. -

Modulation of Mitochondrial DNA Copy Number in a Model of Glioblastoma
Sun and St John Epigenetics & Chromatin (2018) 11:53 https://doi.org/10.1186/s13072-018-0223-z Epigenetics & Chromatin RESEARCH Open Access Modulation of mitochondrial DNA copy number in a model of glioblastoma induces changes to DNA methylation and gene expression of the nuclear genome in tumours Xin Sun1,2 and Justin C. St John1,2* Abstract Background: There are multiple copies of mitochondrial DNA (mtDNA) present in each cell type, and they are strictly regulated in a cell-specifc manner by a group of nuclear-encoded mtDNA-specifc replication factors. This strict regu- lation of mtDNA copy number is mediated by cell-specifc DNA methylation of these replication factors. Glioblastoma multiforme, HSR-GBM1, cells are hyper-methylated and maintain low mtDNA copy number to support their tumori- genic status. We have previously shown that when HSR-GBM1 cells with 50% of their original mtDNA content were inoculated into mice, tumours grew more aggressively than non-depleted cells. However, when the cells possessed only 3% and 0.2% of their original mtDNA content, tumour formation was less frequent and the initiation of tumori- genesis was signifcantly delayed. Importantly, the process of tumorigenesis was dependent on mtDNA copy number being restored to pre-depletion levels. Results: By performing whole genome MeDIP-Seq and RNA-Seq on tumours generated from cells possessing 100%, 50%, 0.3% and 0.2% of their original mtDNA content, we determined that restoration of mtDNA copy number caused signifcant changes to both the nuclear methylome and its transcriptome for each tumour type. The afected genes were specifcally associated with gene networks and pathways involving behaviour, nervous system development, cell diferentiation and regulation of transcription and cellular processes. -

Perspectives in Diabetes Uncoupling Proteins 2 and 3 Potential Regulators of Mitochondrial Energy Metabolism Olivier Boss, Thilo Hagen, and Bradford B
Perspectives in Diabetes Uncoupling Proteins 2 and 3 Potential Regulators of Mitochondrial Energy Metabolism Olivier Boss, Thilo Hagen, and Bradford B. Lowell Mitochondria use energy derived from fuel combustion fuels and oxygen are converted into carbon dioxide, water, to create a proton electrochemical gradient across the and ATP (Fig. 1). The key challenge for the organism is to reg- mitochondrial inner membrane. This intermediate form ulate these many steps so that rates of ATP production are of energy is then used by ATP synthase to synthesize equal to rates of ATP utilization. This is not a small task given AT P. Uncoupling protein-1 (UCP1) is a brown fat–spe- that rates of ATP utilization can quickly increase severalfold cific mitochondrial inner membrane protein with proton (up to 100-fold in muscle during contraction). transport activity. UCP1 catalyzes a highly regulated proton leak, converting energy stored within the mito- Fuel metabolism and oxidative phosphorylation consist chondrial proton electrochemical potential gradient to of many tightly coupled enzymatic reactions (Fig. 1), which heat. This uncouples fuel oxidation from conversion of are regulated, in part, by ADP availability. Control by ADP is ADP to AT P. In rodents, UCP1 activity and brown fat accounted for by the chemiosmotic hypothesis of Mitchell (1). contribute importantly to whole-body energy expendi- Oxidation of fuels via the electron transport chain generates ture. Recently, two additional mitochondrial carriers a proton electrochemical potential gradient ( µH+) across with high similarity to UCP1 were molecularly cloned. the mitochondrial inner membrane. Protons reenter the In contrast to UCP1, UCP2 is expressed widely, and mitochondrial matrix via ATP synthase (F0F1-A TPase) in a UCP3 is expressed preferentially in skeletal muscle. -

NDUFB6 (NM 182739) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC219791 NDUFB6 (NM_182739) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: NDUFB6 (NM_182739) Human Tagged ORF Clone Tag: Myc-DDK Symbol: NDUFB6 Synonyms: B17; CI Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC219791 representing NM_182739 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGACGGGGTACACTCCGGATGAGAAACTGCGGCTGCAGCAGCTGCGAGAGCTGAGAAGGCGATGGCTGA AGGACCAGGAGCTGAGCCCTCGGGAGCCGGTGCTGCCCCCACAGAAGATGGGGCCTATGGAGAAATTCTG GAATAAATTTTTGGAGAATAAATCCCCTTGGAGGAAAATGGTCCATGGGGTATACAAAAAGAGTATCTTT GTTTTCACTCATGTACTTGTACCTGTCTGGATTATTCATTATTACATGAAGTATCATGTTTCTGGTGATA CAATTCTGGAGACTGGAGAAGTAATTCCACCAATGAAAGAATTTCCTGATCAACATCAT ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT ACAAGGATGACGACGATAAGGTTTAA Protein Sequence: >RC219791 representing NM_182739 Red=Cloning site Green=Tags(s) MTGYTPDEKLRLQQLRELRRRWLKDQELSPREPVLPPQKMGPMEKFWNKFLENKSPWRKMVHGVYKKSIF VFTHVLVPVWIIHYYMKYHVSGDTILETGEVIPPMKEFPDQHH TRTRPLEQKLISEEDLAANDILDYKDDDDKV Restriction Sites: SgfI-MluI This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, -

THE FUNCTIONAL SIGNIFICANCE of MITOCHONDRIAL SUPERCOMPLEXES in C. ELEGANS by WICHIT SUTHAMMARAK Submitted in Partial Fulfillment
THE FUNCTIONAL SIGNIFICANCE OF MITOCHONDRIAL SUPERCOMPLEXES in C. ELEGANS by WICHIT SUTHAMMARAK Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Dissertation Advisor: Drs. Margaret M. Sedensky & Philip G. Morgan Department of Genetics CASE WESTERN RESERVE UNIVERSITY January, 2011 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of _____________________________________________________ candidate for the ______________________degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. Dedicated to my family, my teachers and all of my beloved ones for their love and support ii ACKNOWLEDGEMENTS My advanced academic journey began 5 years ago on the opposite side of the world. I traveled to the United States from Thailand in search of a better understanding of science so that one day I can return to my homeland and apply the knowledge and experience I have gained to improve the lives of those affected by sickness and disease yet unanswered by science. Ultimately, I hoped to make the academic transition into the scholarly community by proving myself through scientific research and understanding so that I can make a meaningful contribution to both the scientific and medical communities. The following dissertation would not have been possible without the help, support, and guidance of a lot of people both near and far. I wish to thank all who have aided me in one way or another on this long yet rewarding journey. My sincerest thanks and appreciation goes to my advisors Philip Morgan and Margaret Sedensky. -
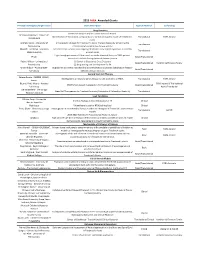
Grantsummaries 2014-2017 Jf
2015 FARA Awarded Grants Principal Investigator/Organization Grant Description Type of Research Co-funding Drug Discovery 2014 Keith Michael Andrus Cardiac Research Award: Veronique Monnier - Université Identification of therapeutic compounds on a cardiac Drosophila model of Friedreich’s Translational FARA Ireland Paris Diderot ataxia Andrew Dancis - University of A therapeutic strategy for Friedreich's ataxia: Frataxin bypass by enhancing the Translational Pennsylvania mitochondrial cysteine desulfurase activity Edward L. Grabczyk - Louisiana Small molecule induced exon skipping of MLH3 to slow repeat expansion in an FRDA Translational State University mousel model High throughput screen of Pfizer small molecule chemical library in FRDA patient- Pfizer Basic/Translational derived cells to look for regulators of frataxin protein Robert Wilson - University of (1) Center of Excellence: Drug Discovery Basic/Translational Hamilton & Finneran Family Pennsylvania (2) Drug testing and development for FA Omar Khdour - Arizona State Approaches to correct mitochondrial abnormalities and synaptic pathology in frataxin- Basic/Translational University deficient sensory neurons Gene & Stem Cell Therapy Helene Puccio - INSERM, IGBMC, Development of neuronal gene therapy for the treatment of FRDA Translational FARA Ireland France Ricardo Pinto Mouro - Harvard FARA Ireland & The National CRISPR/Cas9-based Therapeutics for Friedreich's ataxia Basic/Translational University Ataxia Foundation Joel Gottesfeld - The Scripps Stem Cell Therapeutics for Friedreich's