Functional Reconstitution of Mitochondrial Fe/S Cluster Synthesis on Isu1 Reveals the Involvement of Ferredoxin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dual Role of the Mitochondrial Protein Frataxin in Astrocytic Tumors
Laboratory Investigation (2011) 91, 1766–1776 & 2011 USCAP, Inc All rights reserved 0023-6837/11 $32.00 Dual role of the mitochondrial protein frataxin in astrocytic tumors Elmar Kirches1, Nadine Andrae1, Aline Hoefer2, Barbara Kehler1,3, Kim Zarse4, Martin Leverkus3, Gerburg Keilhoff5, Peter Schonfeld5, Thomas Schneider6, Annette Wilisch-Neumann1 and Christian Mawrin1 The mitochondrial protein frataxin (FXN) is known to be involved in mitochondrial iron homeostasis and iron–sulfur cluster biogenesis. It is discussed to modulate function of the electron transport chain and production of reactive oxygen species (ROS). FXN loss in neurons and heart muscle cells causes an autosomal-dominant mitochondrial disorder, Friedreich’s ataxia. Recently, tumor induction after targeted FXN deletion in liver and reversal of the tumorigenic phenotype of colonic carcinoma cells following FXN overexpression were described in the literature, suggesting a tumor suppressor function. We hypothesized that a partial reversal of the malignant phenotype of glioma cells should occur after FXN transfection, if the mitochondrial protein has tumor suppressor functions in these brain tumors. In astrocytic brain tumors and tumor cell lines, we observed reduced FXN levels compared with non-neoplastic astrocytes. Mitochondrial content (citrate synthase activity) was not significantly altered in U87MG glioblastoma cells stably overexpressing FXN (U87-FXN). Surprisingly, U87-FXN cells exhibited increased cytoplasmic ROS levels, although mitochondrial ROS release was attenuated by FXN, as expected. Higher cytoplasmic ROS levels corresponded to reduced activities of glutathione peroxidase and catalase, and lower glutathione content. The defect of antioxidative capacity resulted in increased susceptibility of U87-FXN cells against oxidative stress induced by H2O2 or buthionine sulfoximine. -

Photosynthetic Metabolism and Nitrogen Reshuffling Are Regulated
plants Article Photosynthetic Metabolism and Nitrogen Reshuffling Are Regulated by Reversible Cysteine Thiol Oxidation Following Nitrogen Deprivation in Chlamydomonas Amanda L. Smythers , Evan W. McConnell , Hailey C. Lewis, Saher N. Mubarek and Leslie M. Hicks * Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA; [email protected] (A.L.S.); [email protected] (E.W.M.); [email protected] (H.C.L.); [email protected] (S.N.M.) * Correspondence: [email protected]; Tel.: +1-919-843-6903 Received: 30 April 2020; Accepted: 19 June 2020; Published: 23 June 2020 Abstract: As global temperatures climb to historic highs, the far-reaching effects of climate change have impacted agricultural nutrient availability. This has extended to low latitude oceans, where a deficit in both nitrogen and phosphorus stores has led to dramatic decreases in carbon sequestration in oceanic phytoplankton. Although Chlamydomonas reinhardtii, a freshwater model green alga, has shown drastic systems-level alterations following nitrogen deprivation, the mechanisms through which these alterations are triggered and regulated are not fully understood. This study examined the role of reversible oxidative signaling in the nitrogen stress response of C. reinhardtii. Using oxidized cysteine resin-assisted capture enrichment coupled with label-free quantitative proteomics, 7889 unique oxidized cysteine thiol identifiers were quantified, with 231 significantly changing peptides from 184 proteins following 2 h of nitrogen deprivation. These results demonstrate that the cellular response to nitrogen assimilation, photosynthesis, pigment biosynthesis, and lipid metabolism are regulated by reversible oxidation. An enhanced role of non-damaging oxidative pathways is observed throughout the photosynthetic apparatus that provides a framework for further analysis in phototrophs. -

The Pathogenetic Role of Β-Cell Mitochondria in Type 2 Diabetes
236 3 Journal of M Fex et al. Mitochondria in β-cells 236:3 R145–R159 Endocrinology REVIEW The pathogenetic role of β-cell mitochondria in type 2 diabetes Malin Fex1, Lisa M Nicholas1, Neelanjan Vishnu1, Anya Medina1, Vladimir V Sharoyko1, David G Nicholls1, Peter Spégel1,2 and Hindrik Mulder1 1Department of Clinical Sciences in Malmö, Unit of Molecular Metabolism, Lund University Diabetes Centre, Clinical Research Center, Malmö University Hospital, Lund University, Malmö, Sweden 2Department of Chemistry, Center for Analysis and Synthesis, Lund University, Sweden Correspondence should be addressed to H Mulder: [email protected] Abstract Mitochondrial metabolism is a major determinant of insulin secretion from pancreatic Key Words β-cells. Type 2 diabetes evolves when β-cells fail to release appropriate amounts f TCA cycle of insulin in response to glucose. This results in hyperglycemia and metabolic f coupling signal dysregulation. Evidence has recently been mounting that mitochondrial dysfunction f oxidative phosphorylation plays an important role in these processes. Monogenic dysfunction of mitochondria is a f mitochondrial transcription rare condition but causes a type 2 diabetes-like syndrome owing to β-cell failure. Here, f genetic variation we describe novel advances in research on mitochondrial dysfunction in the β-cell in type 2 diabetes, with a focus on human studies. Relevant studies in animal and cell models of the disease are described. Transcriptional and translational regulation in mitochondria are particularly emphasized. The role of metabolic enzymes and pathways and their impact on β-cell function in type 2 diabetes pathophysiology are discussed. The role of genetic variation in mitochondrial function leading to type 2 diabetes is highlighted. -

Src Inhibitors Modulate Frataxin Protein Levels
Human Molecular Genetics, 2015, Vol. 24, No. 15 4296–4305 doi: 10.1093/hmg/ddv162 Advance Access Publication Date: 6 May 2015 Original Article ORIGINAL ARTICLE Src inhibitors modulate frataxin protein levels Downloaded from https://academic.oup.com/hmg/article/24/15/4296/2453025 by guest on 28 September 2021 Fabio Cherubini1, Dario Serio1, Ilaria Guccini1, Silvia Fortuni1,2, Gaetano Arcuri1, Ivano Condò1, Alessandra Rufini1,2, Shadman Moiz1, Serena Camerini3, Marco Crescenzi3, Roberto Testi1,2 and Florence Malisan1,* 1Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy, 2Fratagene Therapeutics Ltd, 22 Northumberland Rd, Dublin, Ireland and 3Department of Cell Biology and Neurosciences, Italian National Institute of Health, Viale Regina Elena, 299, 00161 Rome, Italy *To whom correspondence should be addressed at: Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy. Tel: +39 0672596501; Fax: +39 0672596505; Email: [email protected] Abstract Defective expression of frataxin is responsible for the inherited, progressive degenerative disease Friedreich’s Ataxia (FRDA). There is currently no effective approved treatment for FRDA and patients die prematurely. Defective frataxin expression causes critical metabolic changes, including redox imbalance and ATP deficiency. As these alterations are known to regulate the tyrosine kinase Src, we investigated whether Src might in turn affect frataxin expression. We found that frataxin can be phosphorylated by Src. Phosphorylation occurs primarily on Y118 and promotes frataxin ubiquitination, a signal for degradation. Accordingly, Src inhibitors induce accumulation of frataxin but are ineffective on a non-phosphorylatable frataxin- Y118F mutant. -

Sex Differences in the Genetic Architecture of Depression
www.nature.com/scientificreports OPEN Sex diferences in the genetic architecture of depression Hee-Ju Kang1,3, Yoomi Park2,3, Kyung-Hun Yoo2, Ki-Tae Kim2, Eun-Song Kim1, Ju-Wan Kim1, Sung-Wan Kim1, Il-Seon Shin1, Jin-Sang Yoon1, Ju Han Kim2,3 ✉ & Jae-Min Kim1,3 ✉ The prevalence and clinical characteristics of depressive disorders difer between women and men; however, the genetic contribution to sex diferences in depressive disorders has not been elucidated. To evaluate sex-specifc diferences in the genetic architecture of depression, whole exome sequencing of samples from 1000 patients (70.7% female) with depressive disorder was conducted. Control data from healthy individuals with no psychiatric disorder (n = 72, 26.4% female) and East-Asian subpopulation 1000 Genome Project data (n = 207, 50.7% female) were included. The genetic variation between men and women was directly compared using both qualitative and quantitative research designs. Qualitative analysis identifed fve genetic markers potentially associated with increased risk of depressive disorder in females, including three variants (rs201432982 within PDE4A, and rs62640397 and rs79442975 within FDX1L) mapping to chromosome 19p13.2 and two novel variants (rs820182 and rs820148) within MYO15B at the chromosome 17p25.1 locus. Depressed patients homozygous for these variants showed more severe depressive symptoms and higher suicidality than those who were not homozygotes (i.e., heterozygotes and homozygotes for the non-associated allele). Quantitative analysis demonstrated that the genetic burden of protein-truncating and deleterious variants was higher in males than females, even after permutation testing. Our study provides novel genetic evidence that the higher prevalence of depressive disorders in women may be attributable to inherited variants. -
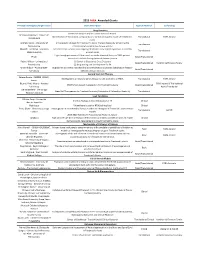
Grantsummaries 2014-2017 Jf
2015 FARA Awarded Grants Principal Investigator/Organization Grant Description Type of Research Co-funding Drug Discovery 2014 Keith Michael Andrus Cardiac Research Award: Veronique Monnier - Université Identification of therapeutic compounds on a cardiac Drosophila model of Friedreich’s Translational FARA Ireland Paris Diderot ataxia Andrew Dancis - University of A therapeutic strategy for Friedreich's ataxia: Frataxin bypass by enhancing the Translational Pennsylvania mitochondrial cysteine desulfurase activity Edward L. Grabczyk - Louisiana Small molecule induced exon skipping of MLH3 to slow repeat expansion in an FRDA Translational State University mousel model High throughput screen of Pfizer small molecule chemical library in FRDA patient- Pfizer Basic/Translational derived cells to look for regulators of frataxin protein Robert Wilson - University of (1) Center of Excellence: Drug Discovery Basic/Translational Hamilton & Finneran Family Pennsylvania (2) Drug testing and development for FA Omar Khdour - Arizona State Approaches to correct mitochondrial abnormalities and synaptic pathology in frataxin- Basic/Translational University deficient sensory neurons Gene & Stem Cell Therapy Helene Puccio - INSERM, IGBMC, Development of neuronal gene therapy for the treatment of FRDA Translational FARA Ireland France Ricardo Pinto Mouro - Harvard FARA Ireland & The National CRISPR/Cas9-based Therapeutics for Friedreich's ataxia Basic/Translational University Ataxia Foundation Joel Gottesfeld - The Scripps Stem Cell Therapeutics for Friedreich's -

A Novel Complex Neurological Phenotype Due to a Homozygous Mutation in FDX2
doi:10.1093/brain/awy172 BRAIN 2018: 141; 2289–2298 | 2289 Downloaded from https://academic.oup.com/brain/article-abstract/141/8/2289/5054138 by FAC.MED.RIB.PRETO-BIBL.CENTRAL-USP user on 26 October 2018 A novel complex neurological phenotype due to a homozygous mutation in FDX2 Juliana Gurgel-Giannetti,1,* David S. Lynch,2,3,* Anderson Rodrigues Branda˜o de Paiva,4 Leandro Tavares Lucato,5 Guilherme Yamamoto,6 Christer Thomsen,7 Somsuvro Basu,8 Fernando Freua,4 Alexandre Varella Giannetti,9 Bruno Della Ripa de Assis,4 Mara Dell Ospedale Ribeiro,4 Isabella Barcelos,4 Katiane Saya˜o Souza,4 Fernanda Monti,4 Uira´ Souto Melo,6 Simone Amorim,4 Leonardo G. L. Silva,6 Lu´cia Ineˆs Macedo-Souza,6 Angela M. Vianna-Morgante,6 Michio Hirano,10 Marjo S. Van der Knaap,11 Roland Lill,8,12 Mariz Vainzof,6 Anders Oldfors,7 Henry Houlden2,3 and Fernando Kok4,6 *These authors contributed equally to this work. Defects in iron–sulphur [Fe-S] cluster biogenesis are increasingly recognized as causing neurological disease. Mutations in a number of genes that encode proteins involved in mitochondrial [Fe-S] protein assembly lead to complex neurological phenotypes. One class of proteins essential in the early cluster assembly are ferredoxins. FDX2 is ubiquitously expressed and is essential in the de novo formation of [2Fe-2S] clusters in humans. We describe and genetically define a novel complex neurological syndrome identified in two Brazilian families, with a novel homozygous mutation in FDX2. Patients were clinically evaluated, underwent MRI, nerve conduction studies, EMG and muscle biopsy. -

Bacteriostatic and Bactericidal Effects of Free Nitrous Acid on Model Microbes in Wastewater Treatment
Bacteriostatic and bactericidal effects of free nitrous acid on model microbes in wastewater treatment Shuhong Gao Master of Science Harbin Institute of Technology, Harbin, China A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2016 School of Chemical Engineering Advanced Water Management Centre Abstract There is great potential to use free nitrous acid (FNA), the protonated form of nitrite (HNO2), as an antimicrobial agent due to its bacteriostatic and bactericidal effects on a range of microorganisms. However, the antimicrobial mechanism of FNA is largely unknown. The overall objective of this thesis is to elucidate the responses of two model bacteria, namely Psuedomonas aeruginosa PAO1 and Desulfovibrio vulgaris Hildenborough, in wastewater treatment in terms of microbial susceptibility, tolerance and resistance to FNA exposure. The effects of FNA on the opportunistic pathogen P. aeruginosa PAO1, a well-studied denitrifier capable of nitrate/nitrite reduction through anaerobic respiration, were determined. It was revealed that the antimicrobial effect of FNA is concentration-determined and population-specific. By applying different levels of FNA, it was seen that 0.1 to 0.2 mg N/L FNA exerted a temporary inhibitory effect on P. aeruginosa PAO1 growth, while complete respiratory growth inhibition was not detected until an FNA concentration of 1.0 mg N/L was applied. The FNA concentration of 5.0 mg N/L caused complete cell killing and likely cell lysis. Differential killing by FNA in the P. aeruginosa PAO1 subpopulations was detected, suggesting intra-strain heterogeneity. A delayed recovery from FNA treatment suggested that FNA caused cell damage which required repair prior to P. -

Structure and Functional Dynamics of the Mitochondrial Fe/S Cluster Synthesis Complex
ARTICLE DOI: 10.1038/s41467-017-01497-1 OPEN Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex Michal T. Boniecki1, Sven A. Freibert 2, Ulrich Mühlenhoff2, Roland Lill 2,3 & Miroslaw Cygler 1,4 Iron–sulfur (Fe/S) clusters are essential protein cofactors crucial for many cellular functions including DNA maintenance, protein translation, and energy conversion. De novo Fe/S cluster synthesis occurs on the mitochondrial scaffold protein ISCU and requires cysteine 1234567890 desulfurase NFS1, ferredoxin, frataxin, and the small factors ISD11 and ACP (acyl carrier protein). Both the mechanism of Fe/S cluster synthesis and function of ISD11-ACP are poorly understood. Here, we present crystal structures of three different NFS1-ISD11-ACP complexes with and without ISCU, and we use SAXS analyses to define the 3D architecture of the complete mitochondrial Fe/S cluster biosynthetic complex. Our structural and biochemical studies provide mechanistic insights into Fe/S cluster synthesis at the catalytic center defined by the active-site Cys of NFS1 and conserved Cys, Asp, and His residues of ISCU. We assign specific regulatory rather than catalytic roles to ISD11-ACP that link Fe/S cluster synthesis with mitochondrial lipid synthesis and cellular energy status. 1 Department of Biochemistry, University of Saskatchewan, 107 Wiggins Road, Saskatoon, SK, Canada S7N 5E5. 2 Institut für Zytobiologie und Zytopathologie, Philipps-Universität, Robert-Koch-Strasse 6, 35032 Marburg, Germany. 3 LOEWE Zentrum für Synthetische Mikrobiologie SynMikro, Hans- Meerwein-Strasse, 35043 Marburg, Germany. 4 Department of Biochemistry, McGill University, 3649 Promenade Sir William Osler, Montreal, QC, Canada H3G 0B1. -

Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’S Disease
biomedicines Review Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease Heng-Chung Kung 1,2 , Kai-Jung Lin 1,3 , Chia-Te Kung 4,* and Tsu-Kung Lin 1,5,6,* 1 Center for Mitochondrial Research and Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan; [email protected] (H.-C.K.); [email protected] (K.-J.L.) 2 Department of Biology, Krieger School of Arts and Sciences, Johns Hopkins University, Baltimore, MD 21218, USA 3 Department of Family Medicine, National Taiwan University Hospital, Taipei 100225, Taiwan 4 Department of Emergency Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan 5 Department of Neurology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan 6 Center of Parkinson’s Disease, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan * Correspondence: [email protected] (C.-T.K.); [email protected] (T.-K.L.) Abstract: Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by dopaminergic neuronal loss. The exact pathogenesis of PD is complex and not yet completely understood, but research has established the critical role mitochondrial dysfunction plays in the development of PD. As the main producer of cytosolic reactive oxygen species (ROS), mito- chondria are particularly susceptible to oxidative stress once an imbalance between ROS generation and the organelle’s antioxidative system occurs. An overabundance of ROS in the mitochondria can Citation: Kung, H.-C.; Lin, K.-J.; lead to mitochondrial dysfunction and further vicious cycles. -

Strand Breaks for P53 Exon 6 and 8 Among Different Time Course of Folate Depletion Or Repletion in the Rectosigmoid Mucosa
SUPPLEMENTAL FIGURE COLON p53 EXONIC STRAND BREAKS DURING FOLATE DEPLETION-REPLETION INTERVENTION Supplemental Figure Legend Strand breaks for p53 exon 6 and 8 among different time course of folate depletion or repletion in the rectosigmoid mucosa. The input of DNA was controlled by GAPDH. The data is shown as ΔCt after normalized to GAPDH. The higher ΔCt the more strand breaks. The P value is shown in the figure. SUPPLEMENT S1 Genes that were significantly UPREGULATED after folate intervention (by unadjusted paired t-test), list is sorted by P value Gene Symbol Nucleotide P VALUE Description OLFM4 NM_006418 0.0000 Homo sapiens differentially expressed in hematopoietic lineages (GW112) mRNA. FMR1NB NM_152578 0.0000 Homo sapiens hypothetical protein FLJ25736 (FLJ25736) mRNA. IFI6 NM_002038 0.0001 Homo sapiens interferon alpha-inducible protein (clone IFI-6-16) (G1P3) transcript variant 1 mRNA. Homo sapiens UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 15 GALNTL5 NM_145292 0.0001 (GALNT15) mRNA. STIM2 NM_020860 0.0001 Homo sapiens stromal interaction molecule 2 (STIM2) mRNA. ZNF645 NM_152577 0.0002 Homo sapiens hypothetical protein FLJ25735 (FLJ25735) mRNA. ATP12A NM_001676 0.0002 Homo sapiens ATPase H+/K+ transporting nongastric alpha polypeptide (ATP12A) mRNA. U1SNRNPBP NM_007020 0.0003 Homo sapiens U1-snRNP binding protein homolog (U1SNRNPBP) transcript variant 1 mRNA. RNF125 NM_017831 0.0004 Homo sapiens ring finger protein 125 (RNF125) mRNA. FMNL1 NM_005892 0.0004 Homo sapiens formin-like (FMNL) mRNA. ISG15 NM_005101 0.0005 Homo sapiens interferon alpha-inducible protein (clone IFI-15K) (G1P2) mRNA. SLC6A14 NM_007231 0.0005 Homo sapiens solute carrier family 6 (neurotransmitter transporter) member 14 (SLC6A14) mRNA. -

Stríbrná and Julius Lukes Hassan Hashimi, Lindsay Mcdonald, Eva
Bioenergetics: Trypanosome Letm1 Protein Is Essential for Mitochondrial Potassium Homeostasis Hassan Hashimi, Lindsay McDonald, Eva Stríbrná and Julius Lukes J. Biol. Chem. 2013, 288:26914-26925. doi: 10.1074/jbc.M113.495119 originally published online July 26, 2013 Access the most updated version of this article at doi: 10.1074/jbc.M113.495119 Find articles, minireviews, Reflections and Classics on similar topics on the JBC Affinity Sites. Alerts: • When this article is cited • When a correction for this article is posted Click here to choose from all of JBC's e-mail alerts Supplemental material: http://www.jbc.org/content/suppl/2013/07/26/M113.495119.DC1.html This article cites 60 references, 26 of which can be accessed free at http://www.jbc.org/content/288/37/26914.full.html#ref-list-1 Downloaded from http://www.jbc.org/ at Edinburgh University Library on September 13, 2013 THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 288, NO. 37, pp. 26914–26925, September 13, 2013 © 2013 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A. Trypanosome Letm1 Protein Is Essential for Mitochondrial Potassium Homeostasis*□S Received for publication, June 19, 2013, and in revised form, July 23, 2013 Published, JBC Papers in Press, July 26, 2013, DOI 10.1074/jbc.M113.495119 Hassan Hashimi‡§1, Lindsay McDonald‡2, Eva Strˇíbrná‡, and Julius Lukesˇ‡§3 From the ‡Institute of Parasitology, Biology Centre, Czech Academy of Sciences and the §Faculty of Science, University of South Bohemia, 370 05 Cˇeské Budeˇjovice (Budweis), Czech Republic Background: Letm1 is a mitochondrial protein attributed disparate roles, including cation/proton antiport and translation.