Analysis of the Primary Sequence and Secondary Structure of the Unusually Long SSU Rrna of the Soil Bug, Armadillidium Vulgare
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

What Are Their Roles in Mitochondrial Protein Synthesis?
Characterisation of human mtRF1 and C12orf65: What are their roles in mitochondrial protein synthesis? Aleksandra Pajak M.Res Thesis submitted to Newcastle University in candidature for the degree of Doctor of Philosophy Newcastle University Faculty of Medical Sciences Institute for Ageing and Health Mitochondrial Research Group January 2013 Abstract Mitochondria have their own protein synthesis machinery that synthesises the oxidative phosphorylation components encoded by their mtDNA. This translation process consists of four main phases: initiation, elongation, termination and ribosome recycling. Termination and its control have been the least investigated. Recently, however, the termination factor, mtRF1a, has been characterised as sufficient to release all the nascent proteins from the mitoribosome. Furthermore, bioinformatics has identified three additional members of this mitochondrial release factor family namely, mtRF1, C12orf65 and ICT1. The latter is now known to be incorporated into the mitoribosome but its exact function remains unclear. My project has therefore focussed on characterising the remaining two factors; mtRF1 and C12orf65, and investigating their possible involvement in mitochondrial protein synthesis. It has been demonstrated that protein synthesis is not perfect and bacterial ribosomes not infrequently stall during translation. This can result from limiting amounts of charged tRNAs, stable secondary structures, or truncated/degraded transcripts. Ribosome stalling has been shown to cause growth arrest. In order to prevent that and maintain high efficiency of mitochondrial protein synthesis such stalled complexes need to be rapidly recycled. Bacteria have developed at least three distinct mechanisms by which ribosomes can be rescued. Contrastingly, despite the presence of truncated mRNAs in mitochondria, no such quality control mechanisms have been identified in these organelles. -

Analysis of the Relationship Between Ribosomal Protein and SSU Processome Assembly in Saccharomyces Cerevisiae
Analysis of the relationship between ribosomal protein and SSU processome assembly in Saccharomyces cerevisiae Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) der naturwissenschaftlichen Fakultät III – Biologie und vorklinische Medizin - der Universität Regensburg vorgelegt von Steffen Jakob aus Wolfen Januar 2010 Promotionsgesuch eingereicht am: 13. Januar 2010 Die Arbeit wurde angeleitet von: Prof. Dr. Herbert Tschochner Prüfungsausschuss: Vorsitzender: Prof. Dr. Armin Kurtz 1. Prüfer: Prof. Dr. Herbert Tschochner 2. Prüfer: Prof. Dr. Rainer Deutzmann 3. Prüfer: Prof. Dr. Wolfgang Seufert Tag der mündlichen Prüfung: 24. März 2010 Die vorliegende Arbeit wurde in der Zeit von April 2006 bis Januar 2010 am Lehrstuhl Biochemie III des Institutes für Biochemie, Genetik und Mikrobiologie der Naturwissenschaftlichen Fakultät III der Universität zu Regensburg unter Anleitung von Dr. Philipp Milkereit im Labor von Prof. Dr. Herbert Tschochner angefertigt. Ich erkläre hiermit, dass ich diese Arbeit selbst verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel verwendet habe. Diese Arbeit war bisher noch nicht Bestandteil eines Prüfungsverfahrens. Andere Promotionsversuche wurden nicht unternommen. Regensburg, den 13. Januar 2010 Steffen Jakob Table of Contents Table of Contents 1 SUMMARY ...................................................................................................... 1 2 INTRODUCTION ............................................................................................ -
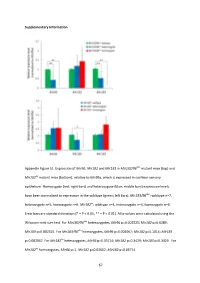
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Micrornas Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins
cells Review microRNAs Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins Abu Musa Md Talimur Reza 1,2 and Yu-Guo Yuan 1,3,* 1 Jiangsu Co-Innovation Center of Prevention and Control of Important Animal Infectious Diseases and Zoonoses, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China; [email protected] 2 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawi´nskiego5a, 02-106 Warsaw, Poland 3 Jiangsu Key Laboratory of Zoonosis/Joint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China * Correspondence: [email protected]; Tel.: +86-514-8797-9228 Abstract: Ribosomal proteins (RPs) are mostly derived from the energy-consuming enzyme families such as ATP-dependent RNA helicases, AAA-ATPases, GTPases and kinases, and are important structural components of the ribosome, which is a supramolecular ribonucleoprotein complex, composed of Ribosomal RNA (rRNA) and RPs, coordinates the translation and synthesis of proteins with the help of transfer RNA (tRNA) and other factors. Not all RPs are indispensable; in other words, the ribosome could be functional and could continue the translation of proteins instead of lacking in some of the RPs. However, the lack of many RPs could result in severe defects in the biogenesis of ribosomes, which could directly influence the overall translation processes and global expression of the proteins leading to the emergence of different diseases including cancer. While microRNAs (miRNAs) are small non-coding RNAs and one of the potent regulators of the post-transcriptional 0 gene expression, miRNAs regulate gene expression by targeting the 3 untranslated region and/or coding region of the messenger RNAs (mRNAs), and by interacting with the 50 untranslated region, Citation: Reza, A.M.M.T.; Yuan, Y.-G. -

History of the Ribosome and the Origin of Translation
History of the ribosome and the origin of translation Anton S. Petrova,1, Burak Gulena, Ashlyn M. Norrisa, Nicholas A. Kovacsa, Chad R. Berniera, Kathryn A. Laniera, George E. Foxb, Stephen C. Harveyc, Roger M. Wartellc, Nicholas V. Huda, and Loren Dean Williamsa,1 aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332; bDepartment of Biology and Biochemistry, University of Houston, Houston, TX, 77204; and cSchool of Biology, Georgia Institute of Technology, Atlanta, GA 30332 Edited by David M. Hillis, The University of Texas at Austin, Austin, TX, and approved November 6, 2015 (received for review May 18, 2015) We present a molecular-level model for the origin and evolution of building up of the functional centers, proceeds to the establishment the translation system, using a 3D comparative method. In this model, of the common core, and continues to the development of large the ribosome evolved by accretion, recursively adding expansion metazoan rRNAs. segments, iteratively growing, subsuming, and freezing the rRNA. Incremental evolution of function is mapped out by stepwise Functions of expansion segments in the ancestral ribosome are accretion of rRNA. In the extant ribosome, specific segments of assigned by correspondence with their functions in the extant rRNA perform specific functions including peptidyl transfer, ribosome. The model explains the evolution of the large ribosomal subunit association, decoding, and energy-driven translocation subunit, the small ribosomal subunit, tRNA, and mRNA. Prokaryotic (11). The model assumes that the correlations of rRNA segments ribosomes evolved in six phases, sequentially acquiring capabilities with their functions have been reasonably maintained over the for RNA folding, catalysis, subunit association, correlated evolution, broad course of ribosomal evolution. -

HUMAN RIBOSOME BIOGENESIS and the REGULATION of the TUMOUR SUPPRESSOR P53
HUMAN RIBOSOME BIOGENESIS AND THE REGULATION OF THE TUMOUR SUPPRESSOR p53 Andria Pelava Submitted for Doctor of Philosophy Final submission: December 2016 Institute of Cell and Molecular Biosciences Faculty of Medical Sciences Newcastle University ii Abstract Ribosome production is an energetically expensive and, therefore, highly regulated process. Defects in ribosome biogenesis lead to genetic diseases called Ribosomopathies, such as Dyskeratosis Congenita (DC), and mutations in ribosomal proteins and ribosome biogenesis factors are linked to multiple types of cancer. During ribosome biogenesis, the ribosomal RNAs (rRNAs) are processed and modified, and defects in ribosome biogenesis lead to the activation of p53. This project aimed to investigate the functions of Dyskerin, mutated in X-linked DC, in human ribosome biogenesis and p53 regulation, and to explore the link between ribosome production and p53 homeostasis. Dyskerin is an rRNA pseudouridine synthase and required for telomere maintenance. There is some debate as to whether DC is the result of telomere maintenance or ribosome biogenesis defects. It is shown here that human Dyskerin is required for the production of both LSU and SSU, and knockdown of Dyskerin leads to p53 activation via inhibition of MDM2 via the 5S RNP, an LSU assembly intermediate which accumulates after ribosome biogenesis defects. My data indicate that p53 activation, due to defects in ribosome biogenesis, may contribute to the clinical symptoms seen in patients suffering with DC. In addition, it is shown that defects in early or late stages of SSU or LSU biogenesis, result in activation of p53 via the 5S RNP-MDM2 pathway, and that p53 is induced in less than 12 hours after ribosome biogenesis defects. -

Colony PCR Amplification of the 16S Ribosomal RNA Gene I. OBJECTIVES
Lab: Colony PCR amplification of the 16S ribosomal RNA gene I. OBJECTIVES (1) Learn how to use the polymerase chain reaction (PCR) to amplify the small subunit ribosomal RNA (SSU rRNA) gene from a bacterial colony; (2) Learn how to run an agarose gel to visualize the resulting PCR amplicons and extract the amplified DNA from the agarose gel. II. INTRODUCTION Microbial community structure is a critical determinant of the biogeochemical processes occurring within the pelagic marine ecosystem. Due to the difficulties of cultivating many marine microbes, microbial ecologists frequently employ molecular biology-based techniques to reveal the complexity of aquatic microbial communities. Culture-independent techniques for characterizing microbial biodiversity are primarily based on the analysis of small subunit ribosomal RNA (SSU rRNA) genes from environmental samples (e.g. Giovannoni et al. 1990). This gene is an excellent phylogenetic marker for Bacteria and Archaea. With the aid of the polymerase chain reaction (PCR), these strategies have greatly enhanced our ability to describe the genetic diversity of microorganisms in the natural environment without the need for cultivation. The cloning and sequencing of PCR-amplified SSU rRNA genes is now a routine procedure. An overview of the typical steps is as follows: 1) SSU rDNAs (rDNA = rRNA gene) are amplified via the polymerase chain reaction using conserved oligonucleotide primers, 2) amplified products are cloned into a plasmid vector, 3) E. coli cell are transformed with the recombinant vectors 4) individual colonies of transformed E. coli are picked and the plasmids are purified 5) the purified plasmids are used as a template for the sequencing reaction. -

Ribosomal DNA Copy Loss and Repeat Instability in ATRX-Mutated Cancers
Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers Maheshi Udugamaa,1, Elaine Sanijb,c,1, Hsiao P. J. Voona, Jinbae Sonb, Linda Hiia, Jeremy D. Hensond, F. Lyn Chana, Fiona T. M. Changa, Yumei Liue, Richard B. Pearsona,b,f,g, Paul Kalitsish, Jeffrey R. Manni, Philippe Collasj,k, Ross D. Hannana,b,f,g,l, and Lee H. Wonga,2 aDepartment of Biochemistry and Molecular Biology, Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia; bResearch Division, Peter MacCallum Cancer Centre, Parkville, VIC 2010, Australia; cDepartment of Pathology, The University of Melbourne, Parkville, VIC 3010, Australia; dCancer Cell Immortality Group, Adult Cancer Program, Prince of Wales Clinical School, University of New South Wales, Randwick, NSW 2052, Australia; eCollege of Animal Science and Technology, Henan University of Science and Technology, Luoyang, Henan Province, 471023, China; fSir Peter MacCallum Department of Oncology, The University of Melbourne, VIC 3010, Australia; gDepartment of Biochemistry and Molecular Biology, The University of Melbourne, Parkville, VIC 3010, Australia; hDepartment of Paediatrics, Murdoch Children’s Research Institute, Royal Children’s Hospital, University of Melbourne, Parkville, VIC 3052, Australia; iGenome Modification Platform, Monash University, Clayton, VIC 3800, Australia; jDepartment of Molecular Medicine, Institute of Basic Medical Sciences, Faculty of Medicine, University of Oslo, 0317 Oslo, Norway; kNorwegian Center for Stem Cell Research, Department of Immunology -

The Small Subunit of the Mitoribosome from Andalucia Godoyi. Isolation and Study of Its Protein Composition”
UNIVERSITÉ DE MONTRÉAL DÉPARTEMENT DE BIOCHIMIE ET MÉDECINE MOLÉCULAIRE FACULTÉ DE MÉDECINE “The small subunit of the mitoribosome from Andalucia godoyi. Isolation and study of its protein composition” Présenté par José Angel Gonzalez Alcazar Mémoire présenté à la Faculté de médecine en vue de l’obtention du grade de maîtrise en biochimie 20 mars 2018 © José Angel Gonzalez Alcazar, 2018 UNIVERSITÉ DE MONTRÉAL DÉPARTEMENT DE BIOCHIMIE ET MÉDECINE MOLÉCULAIRE FACULTÉ DE MÉDECINE Ce mémoire intitulé: “The small subunit of the mitoribosome from Andalucia godoyi. Isolation and study of its protein composition” Présenté par : José Angel Gonzalez Alcazar a été évalué par un jury composé des personnes suivantes : Dr. Sebastian Pechmann Dre. Gertraud Burger Dr. Martin Schmeing 1 TABLE OF CONTENTS 1. INTRODUCTION ................................................................................................... 10 1.1 Mitochondrial genomes.................................................................................... 10 1.2 The most bacteria-like mtDNAs...................................................................... 11 1.3 Prokaryotic translation .................................................................................... 12 1.4 Translation in mammalian mitochondria ...................................................... 13 1.4 Differences of translation in bacteria, and mammalian and yeast mitochondria ................................................................................................................ 15 1.5 Ribosome -

Characterization of R-Protein Variants in Saccharomyces Cerevisiae
Characterization of r-protein variants in Saccharomyces cerevisiae DISSERTATION ZUR ERLANGUNG DES DOKTORGRADES DER NATURWISSENSCHAFTEN (DR. RER. NAT.) DER NATURWISSENSCHAFTLICHEN FAKULTÄT III BIOLOGIE UND VORKLINISCHE MEDIZIN DER UNIVERSITÄT REGENSBURG vorgelegt von Andreas Neueder aus Bogen Regensburg, 2010 Promotionsgesuch eingereicht am: 13.04.2010 Prüfungausschuss: Vorsitzender: Prof. Dr. A. Kurtz 1. Gutachter: Prof. Dr. H. Tschochner 2. Gutachter: Prof. Dr. R. Sterner 3. Prüfer: Prof. Dr. W. Seufert Ersatzprüfer: Prof. Dr. T. Dresselhaus Diese Arbeit wurde unter der Anleitung von Prof. Dr. Herbert Tschochner und Dr. Philipp Milkereit am Lehrstuhl für Biochemie III des Instituts für Mikrobiologie, Genetik und Biochemie der Universität Regensburg erstellt. Ich erkläre hiermit, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Die aus anderen Quellen direkt oder indirekt übernommenen Daten und Konzepte sind unter Angabe der Quelle gekennzeichnet. Insbesondere habe ich nicht die entgeltliche Hilfe von Vermittlungs- bzw. Beratungsdiensten (Promotionsberater oder andere Personen) in Anspruch genommen. Niemand hat von mir unmittelbar oder mittelbar geldwerte Leistungen für Arbeit erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Die Arbeit wurde bisher weder im In- noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt. Regensburg, den Table of contents Table of contents 1 Introduction -

Intraspecific Variation in Small-Subunit Rrna Sequences in Genbank: Why Single Sequences May Not Adequately Represent Prokaryotic Taxa REBECCA A
INTERNATIONALJOURNAL OF SYSTEMATICBACTERIOLOGY, July 1995, p. 595-599 Vol. 45, No. 3 0020-7713/95/$O4.00 + 0 Copyright 8 1995, International Union of Microbiological Societies Intraspecific Variation in Small-Subunit rRNA Sequences in GenBank: Why Single Sequences May Not Adequately Represent Prokaryotic Taxa REBECCA A. CLAYTON,* GRANGER SUTTON, PAUL S. HINKLE, JR., CAROL BULT, AND CHRIS FIELDS? The Institute for Genomic Research, Gaithersbuq, Maryland 20878 Small-subunitrRNA (SSU rRNA) sequencing is a powerful tool to detect, identify, and classify prokaryotic organisms, and there is currently an explosion of SSU rRNA sequencing in the microbiology community. We report unexpectedly high levels of intraspecific variation (within and between strains) of prokaryote SSU rRNA sequences deposited in GenBank. A total of 82% of the prokaryote species with two published SSU rRNA sequences had more variable positions than a 0.1% random sequencing error would predict, and 48% of these sequence pairs had more variable positions than predicted by a 1.0%random sequencing error. Other sources of sequence variability must account for some of this intraspecific variation. Given these results, phylogenetic studies and biodiversity estimates obtained by using prokaryotic SSU rRNA sequences cannot proceed under the assumption that rRNA sequences of single operons from single isolates adequately represent their taxa. Sequencing SSU rRNA molecules from multiple operons and multiple isolates is highly recommended to obtain meaningful phylogenetic hypotheses, as is careful attention to accurate strain identification. The technological advances that have made sequencing or species are found in the literature, some authors have as- rRNA feasible are revolutionizing prokaryotic systematics sumed that the differences represent errors in laboratory pro- (22).