History of the Ribosome and the Origin of Translation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Different Genetic Mechanisms Mediate Spontaneous Versus UVR-Induced
RESEARCH ARTICLE Different genetic mechanisms mediate spontaneous versus UVR-induced malignant melanoma Blake Ferguson1, Herlina Y Handoko1, Pamela Mukhopadhyay1, Arash Chitsazan1, Lois Balmer2,3, Grant Morahan2, Graeme J Walker1†* 1Drug Discovery Group, QIMR Berghofer Medical Research Institute, Herston, Australia; 2Centre for Diabetes Research, Harry Perkins Institute of Medical Research, Perth, Australia; 3School of Medical and Health Sciences, Edith Cowan University, Joondalup, Australia Abstract Genetic variation conferring resistance and susceptibility to carcinogen-induced tumorigenesis is frequently studied in mice. We have now turned this idea to melanoma using the collaborative cross (CC), a resource of mouse strains designed to discover genes for complex diseases. We studied melanoma-prone transgenic progeny across seventy CC genetic backgrounds. We mapped a strong quantitative trait locus for rapid onset spontaneous melanoma onset to Prkdc, a gene involved in detection and repair of DNA damage. In contrast, rapid onset UVR-induced melanoma was linked to the ribosomal subunit gene Rrp15. Ribosome biogenesis was upregulated in skin shortly after UVR exposure. Mechanistically, variation in the ‘usual suspects’ by which UVR may exacerbate melanoma, defective DNA repair, melanocyte proliferation, or inflammatory cell infiltration, did not explain melanoma susceptibility or resistance across the CC. *For correspondence: [email protected] Instead, events occurring soon after exposure, such as dysregulation of ribosome function, which alters many aspects of cellular metabolism, may be important. † Present address: Experimental DOI: https://doi.org/10.7554/eLife.42424.001 Dermatology Group, The University of Queensland Diamantina Institute, Woolloongabba, Australia Introduction Competing interests: The Cutaneous malignant melanoma (MM) is well known to be associated with high levels of sun expo- authors declare that no sure. -

L-Leucine Increases Translation of RPS14 and LARP1 in Erythroblasts
LETTERS TO THE EDITOR Table 1. Top 20 differentially translated known 5’TOP mRNAs in L-leucine increases translation of RPS14 and LARP1 L-leucine treated erythroblasts from del(5q) myelodysplastic syn- in erythroblasts from del(5q) myelodysplastic drome patients. syndrome patients Genes LogFC of TE in patients z score patients Deletion of the long arm of chromosome 5 [del(5q)] is RPS15 3.55 2.46 the most common cytogenetic abnormality found in the RPS27A 3.48 2.40 1 myelodysplastic syndromes (MDS). Patients with the 5q- RPS25 3.47 2.39 syndrome have macrocytic anemia and the del(5q) as the RPS20 3.43 2.35 sole karyotypic abnormality.1 Haploinsufficiency of the ribosomal protein gene RPS14, mapping to the common- RPL12 3.35 2.29 ly deleted region (CDR) on chromosome 5q,2 underlies PABPC4 3.01 2.01 3 the erythroid defect found in the 5q- syndrome, and is RPS24 2.97 1.98 associated with p53 activation,4-6 a block in the process- ing of pre-ribosomal RNA,3 and deregulation of riboso- RPS3 2.95 1.96 mal- and translation-related genes.7 Defective mRNA EEF2 2.83 1.86 translation represents a potential therapeutic target in the RPS18 2.76 1.80 5q- syndrome and other ribosomopathies, such as RPS26 2.75 1.79 Diamond-Blackfan anemia (DBA).8 Evidence suggests that the translation enhancer L- RPS5 2.69 1.74 leucine may have some efficacy in the treatment of the RPS21 2.64 1.70 8 5q- syndrome and DBA. A DBA patient treated with L- RPS9 2.54 1.62 leucine showed a marked improvement in anemia and 8 EIF3E 2.53 1.61 achieved transfusion independence. -

What Are Their Roles in Mitochondrial Protein Synthesis?
Characterisation of human mtRF1 and C12orf65: What are their roles in mitochondrial protein synthesis? Aleksandra Pajak M.Res Thesis submitted to Newcastle University in candidature for the degree of Doctor of Philosophy Newcastle University Faculty of Medical Sciences Institute for Ageing and Health Mitochondrial Research Group January 2013 Abstract Mitochondria have their own protein synthesis machinery that synthesises the oxidative phosphorylation components encoded by their mtDNA. This translation process consists of four main phases: initiation, elongation, termination and ribosome recycling. Termination and its control have been the least investigated. Recently, however, the termination factor, mtRF1a, has been characterised as sufficient to release all the nascent proteins from the mitoribosome. Furthermore, bioinformatics has identified three additional members of this mitochondrial release factor family namely, mtRF1, C12orf65 and ICT1. The latter is now known to be incorporated into the mitoribosome but its exact function remains unclear. My project has therefore focussed on characterising the remaining two factors; mtRF1 and C12orf65, and investigating their possible involvement in mitochondrial protein synthesis. It has been demonstrated that protein synthesis is not perfect and bacterial ribosomes not infrequently stall during translation. This can result from limiting amounts of charged tRNAs, stable secondary structures, or truncated/degraded transcripts. Ribosome stalling has been shown to cause growth arrest. In order to prevent that and maintain high efficiency of mitochondrial protein synthesis such stalled complexes need to be rapidly recycled. Bacteria have developed at least three distinct mechanisms by which ribosomes can be rescued. Contrastingly, despite the presence of truncated mRNAs in mitochondria, no such quality control mechanisms have been identified in these organelles. -

Evolution of Translation EF-Tu: Trna
University of Illinois at Urbana-Champaign Luthey-Schulten Group NIH Resource for Macromolecular Modeling and Bioinformatics Computational Biophysics Workshop Evolution of Translation EF-Tu: tRNA VMD Developer: John Stone MultiSeq Developers Tutorial Authors Elijah Roberts Ke Chen John Eargle John Eargle Dan Wright Zhaleh Ghaemi Jonathan Lai Zan Luthey-Schulten August 2014 A current version of this tutorial is available at http://www.scs.illinois.edu/~schulten/tutorials/ef-tu CONTENTS 2 Contents 1 Introduction 3 1.1 The Elongation Factor Tu . 3 1.2 Getting Started . 4 1.2.1 Requirements . 4 1.2.2 Copying the tutorial files . 4 1.2.3 Working directory . 4 1.2.4 Preferences . 4 1.3 Configuring BLAST for MultiSeq . 5 2 Comparative Analysis of EF-Tu 5 2.1 Finding archaeal EF1A sequences . 6 2.2 Aligning archaeal sequences and removing redundancy . 8 2.3 Finding bacteria EF-Tu sequences . 11 2.4 Performing ClustalW Multiple Sequence and Profile-Profile Align- ments . 12 2.5 Creating Multiple Sequence with MAFFT . 16 2.6 Conservation of EF-Tu among the Bacteria . 16 2.7 Finding conserved residues across the bacterial and archaeal do- mains . 20 2.8 EF-Tu Interface with the Ribosome . 21 3 Computing a Maximum Likelihood Phylogenetic Tree with RAxML 23 3.1 Load the Phylogenetic Tree into MultiSeq . 25 3.2 Reroot and Manipulate the Phylogenetic Tree . 25 4 MultiSeq TCL Scripting: Genomic Context 27 5 Appendix A 30 5.1 Building a BLAST Database . 30 6 Appendix B 31 6.1 Saving QR subset of alignments in PHYLIP and FASTA format 31 6.2 Calculating Maximum Likelihood Trees with RAxML . -

Dynamics of Translation Can Determine the Spatial Organization Of
Dynamics of translation can determine the spatial organization of membrane-bound proteins and their mRNA Elgin Korkmazhana, Hamid Teimourib,c, Neil Petermanb,c, and Erel Levineb,c,1 aHarvard College, Harvard University, Cambridge, MA 02138; bDepartment of Physics, Harvard University, Cambridge, MA 02138; and cFAS Center for Systems Biology, Harvard University, Cambridge, MA 02138 Edited by William Bialek, Princeton University, Princeton, NJ, and approved October 17, 2017 (received for review January 17, 2017) Unlike most macromolecules that are homogeneously distributed these patterns are determined by the organization of the cod- in the bacterial cell, mRNAs that encode inner-membrane proteins ing sequence, the presence of slow codons, the rate of transla- can be concentrated near the inner membrane. Cotranslational tion initiation, and the availability of auxiliary proteins required insertion of the nascent peptide into the membrane brings the for membrane targeting (referred to as the secretory machinery). translating ribosome and the mRNA close to the membrane. This By calculating the distribution of the number of proteins placed suggests that kinetic properties of translation can determine the together in the membrane, we suggest implications of mRNA spatial organization of these mRNAs and proteins, which can be localization on the organization of proteins on the membrane. modulated through posttranscriptional regulation. Here we use a We thus propose a mechanism for the formation of protein clus- simple stochastic model of translation to characterize the effect of ters in the membrane and investigate its implications on the reg- mRNA properties on the dynamics and statistics of its spatial distri- ulation of their size distribution. -

Analysis of the Relationship Between Ribosomal Protein and SSU Processome Assembly in Saccharomyces Cerevisiae
Analysis of the relationship between ribosomal protein and SSU processome assembly in Saccharomyces cerevisiae Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) der naturwissenschaftlichen Fakultät III – Biologie und vorklinische Medizin - der Universität Regensburg vorgelegt von Steffen Jakob aus Wolfen Januar 2010 Promotionsgesuch eingereicht am: 13. Januar 2010 Die Arbeit wurde angeleitet von: Prof. Dr. Herbert Tschochner Prüfungsausschuss: Vorsitzender: Prof. Dr. Armin Kurtz 1. Prüfer: Prof. Dr. Herbert Tschochner 2. Prüfer: Prof. Dr. Rainer Deutzmann 3. Prüfer: Prof. Dr. Wolfgang Seufert Tag der mündlichen Prüfung: 24. März 2010 Die vorliegende Arbeit wurde in der Zeit von April 2006 bis Januar 2010 am Lehrstuhl Biochemie III des Institutes für Biochemie, Genetik und Mikrobiologie der Naturwissenschaftlichen Fakultät III der Universität zu Regensburg unter Anleitung von Dr. Philipp Milkereit im Labor von Prof. Dr. Herbert Tschochner angefertigt. Ich erkläre hiermit, dass ich diese Arbeit selbst verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel verwendet habe. Diese Arbeit war bisher noch nicht Bestandteil eines Prüfungsverfahrens. Andere Promotionsversuche wurden nicht unternommen. Regensburg, den 13. Januar 2010 Steffen Jakob Table of Contents Table of Contents 1 SUMMARY ...................................................................................................... 1 2 INTRODUCTION ............................................................................................ -

Mito-Cytosolic Translational Balance Increased Cytoprotection And
Graphical Abstract Worms Human cells Mice Mito-cytosolic translational balance Genetically mrps-5 RNAi Mitochondrial Cytosolic ribosomes ribosomes ATF4/atf-5 Doxycycline Pharmacologically Increased cytoprotection and longevity Manuscript A conserved mito-cytosolic translational balance links two longevity pathways Marte Molenaars1*, Georges E. Janssens1*, Evan G. Williams2, Aldo Jongejan3, Jiayi Lan2, Sylvie Rabot4, Fatima Joly4, Perry D. Moerland3, Bauke V. Schomakers1,5, Marco Lezzerini1 Yasmine J. Liu1, Mark A. McCormick6,7, Brian K. Kennedy8,9, Michel van Weeghel1,5, Antoine H.C. van Kampen3, Ruedi Aebersold2,10, Alyson W. MacInnes1, Riekelt H. Houtkooper1,11# 1Laboratory Genetic Metabolic Diseases, Amsterdam UMC, University of Amsterdam, Amsterdam Gastroenterology and Metabolism, Amsterdam Cardiovascular Sciences, Amsterdam, The Netherlands 2Institute of Molecular Systems Biology, ETH Zurich, Zürich, Switzerland 3Bioinformatics Laboratory, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands 4Micalis Institute, INRA, AgroParisTech, Université Paris-Saclay, Jouy-en-Josas, France 5Core Facility Metabolomics, Amsterdam UMC, University of Amsterdam, Amsterdam, the Netherlands. 6 Department of Biochemistry and Molecular Biology, School of Medicine, University of New Mexico Health Sciences Center, Albuquerque, USA 7Autophagy, Inflammation, and Metabolism Center of Biological Research Excellence, University of New Mexico Health Sciences Center, Albuquerque, USA 8Buck Institute for Research on Aging, Novato, USA 9Departments -
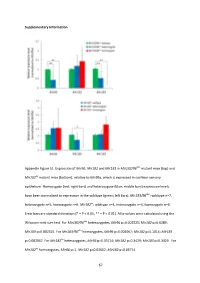
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Mitochondrial Misreading in Skeletal Muscle Accelerates Metabolic Aging and Confers Lipid Accumulation and Increased Inflammation
Downloaded from rnajournal.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press REPORT Mitochondrial misreading in skeletal muscle accelerates metabolic aging and confers lipid accumulation and increased inflammation DIMITRI SHCHERBAKOV,1,4 STEFAN DUSCHA,1,4 REDA JUSKEVICIENE,1 LISA M. RESTELLI,2 STEPHAN FRANK,2 ENDRE LACZKO,3 and ERIK C. BÖTTGER1 1Institut für Medizinische Mikrobiologie, Universität Zürich, 8006 Zürich, Switzerland 2Division of Neuropathology, Institute of Medical Genetics and Pathology, Basel University Hospital, 4031 Basel, Switzerland 3Functional Genomics Center Zurich, ETH Zürich und Universität Zürich, 8057 Zürich, Switzerland ABSTRACT We have recently reported on an experimental model of mitochondrial mistranslation conferred by amino acid exchange V338Y in mitochondrial ribosomal protein MrpS5. Here we used a combination of RNA-seq and metabolic profiling of ho- mozygous transgenic Mrps5V338Y/V338Y mice to analyze the changes associated with the V338Y mutation in postmitotic skeletal muscle. Metabolome analysis demonstrated enhanced levels of age-associated metabolites in the mutant V338Y animals accompanied by increased glycolysis, lipid desaturation and eicosanoid biosynthesis, and alterations of the pentose phosphate pathway. In addition, transcriptome signatures of aged V338Y mutant muscle pointed to elevated inflammation, likely reflecting the increased levels of bioactive lipids. Our findings indicate that mistranslation-mediated impairment of mitochondrial function affects specific bioenergetic processes in muscle in an age-dependent manner. Keywords: mitochondria; misreading; skeletal muscle; aging; metabolome INTRODUCTION express a mtDNA mutator phenotype, with a threefold to fivefold increase in the levels of random point mutations A decline in mitochondrial function has been associated in mtDNA, display respiratory chain dysfunction and fea- with aging and complex age-related changes in metabo- tures of accelerated aging (Trifunovic et al. -

Crystal Structure of the Eukaryotic 60S Ribosomal Subunit in Complex with Initiation Factor 6
Research Collection Doctoral Thesis Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6 Author(s): Voigts-Hoffmann, Felix Publication Date: 2012 Permanent Link: https://doi.org/10.3929/ethz-a-007303759 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library ETH Zurich Dissertation No. 20189 Crystal Structure of the Eukaryotic 60S Ribosomal Subunit in Complex with Initiation Factor 6 A dissertation submitted to ETH ZÜRICH for the degree of Doctor of Sciences (Dr. sc. ETH Zurich) presented by Felix Voigts-Hoffmann MSc Molecular Biotechnology, Universität Heidelberg born April 11, 1981 citizen of Göttingen, Germany accepted on recommendation of Prof. Dr. Nenad Ban (Examiner) Prof. Dr. Raimund Dutzler (Co-examiner) Prof. Dr. Rudolf Glockshuber (Co-examiner) 2012 blank page ii Summary Ribosomes are large complexes of several ribosomal RNAs and dozens of proteins, which catalyze the synthesis of proteins according to the sequence encoded in messenger RNA. Over the last decade, prokaryotic ribosome structures have provided the basis for a mechanistic understanding of protein synthesis. While the core functional centers are conserved in all kingdoms, eukaryotic ribosomes are much larger than archaeal or bacterial ribosomes. Eukaryotic ribosomal rRNA and proteins contain extensions or insertions to the prokaryotic core, and many eukaryotic proteins do not have prokaryotic counterparts. Furthermore, translation regulation and ribosome biogenesis is much more complex in eukaryotes, and defects in components of the translation machinery are associated with human diseases and cancer. -

Nucleolin and Its Role in Ribosomal Biogenesis
NUCLEOLIN: A NUCLEOLAR RNA-BINDING PROTEIN INVOLVED IN RIBOSOME BIOGENESIS Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf vorgelegt von Julia Fremerey aus Hamburg Düsseldorf, April 2016 2 Gedruckt mit der Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf Referent: Prof. Dr. A. Borkhardt Korreferent: Prof. Dr. H. Schwender Tag der mündlichen Prüfung: 20.07.2016 3 Die vorgelegte Arbeit wurde von Juli 2012 bis März 2016 in der Klinik für Kinder- Onkologie, -Hämatologie und Klinische Immunologie des Universitätsklinikums Düsseldorf unter Anleitung von Prof. Dr. A. Borkhardt und in Kooperation mit dem ‚Laboratory of RNA Molecular Biology‘ an der Rockefeller Universität unter Anleitung von Prof. Dr. T. Tuschl angefertigt. 4 Dedicated to my family TABLE OF CONTENTS 5 TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................... 5 LIST OF FIGURES ......................................................................................................10 LIST OF TABLES .......................................................................................................12 ABBREVIATION .........................................................................................................13 ABSTRACT ................................................................................................................19 ZUSAMMENFASSUNG -

USP16 Counteracts Mono-Ubiquitination of Rps27a And
RESEARCH ARTICLE USP16 counteracts mono-ubiquitination of RPS27a and promotes maturation of the 40S ribosomal subunit Christian Montellese1†, Jasmin van den Heuvel1,2, Caroline Ashiono1, Kerstin Do¨ rner1,2, Andre´ Melnik3‡, Stefanie Jonas1§, Ivo Zemp1, Paola Picotti3, Ludovic C Gillet1, Ulrike Kutay1* 1Institute of Biochemistry, ETH Zurich, Zurich, Switzerland; 2Molecular Life Sciences Ph.D. Program, Zurich, Switzerland; 3Institute of Molecular Systems Biology, ETH Zurich, Zurich, Switzerland Abstract Establishment of translational competence represents a decisive cytoplasmic step in the biogenesis of 40S ribosomal subunits. This involves final 18S rRNA processing and release of residual biogenesis factors, including the protein kinase RIOK1. To identify novel proteins promoting the final maturation of human 40S subunits, we characterized pre-ribosomal subunits trapped on RIOK1 by mass spectrometry, and identified the deubiquitinase USP16 among the captured factors. We demonstrate that USP16 constitutes a component of late cytoplasmic pre-40S subunits that promotes the removal of ubiquitin from an internal lysine of ribosomal protein *For correspondence: RPS27a/eS31. USP16 deletion leads to late 40S subunit maturation defects, manifesting in [email protected] incomplete processing of 18S rRNA and retarded recycling of late-acting ribosome biogenesis Present address: †CSL Behring, factors, revealing an unexpected contribution of USP16 to the ultimate step of 40S synthesis. CSL Biologics Research Center, Finally, ubiquitination of RPS27a appears to depend on active translation, pointing at a potential ‡ Bern, Switzerland; MSD Merck connection between 40S maturation and protein synthesis. Sharp & Dohme AG, Lucerne, Switzerland; §Institute of Molecular Biology and Biophysics, ETH Zurich, Zurich, Switzerland Introduction Ribosomes stand at the center of translation in all kingdoms of life, catalyzing the synthesis of pro- Competing interests: The teins by reading a messenger RNA (mRNA) template.