Identification of 40S Ribosomal Protein S8 As a Novel Biomarker for Alcohol‑Associated Hepatocellular Carcinoma Using Weighted Gene Co‑Expression Network Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

C8cc08685k1.Pdf
Electronic Supplementary Material (ESI) for ChemComm. This journal is © The Royal Society of Chemistry 2018 Supporting Information Minimalist Linkers Suitable for Irreversible Inhibitors in Simultaneous Proteome Profiling, Live-Cell Imaging and Drug Screening Cuiping Guo,Yu Chang, Xin Wang, Chengqian Zhang, Piliang Hao*, Ke Ding and Zhengqiu Li* School of Pharmacy, Jinan University, Guangzhou, China 510632 *Corresponding author ([email protected]) 1. General Information All chemicals were purchased from commercial vendors and used without further purification, unless indicated otherwise. All reactions requiring anhydrous conditions were carried out under argon or nitrogen atmosphere using oven-dried glassware. AR-grade solvents were used for all reactions. Reaction progress was monitored by TLC on pre-coated silica plates (Merck 60 F254 nm, 0.25 µm) and spots were visualized by UV, iodine or other suitable stains. Flash column chromatography was carried out using silica gel (Qingdao Ocean). All NMR spectra (1H-NMR, 13C-NMR) were recorded on Bruker 300 MHz/400 MHz NMR spectrometers. Chemical shifts were reported in parts per million (ppm) referenced with respect to appropriate internal standards or residual solvent peaks (CDCl3 = 7.26 ppm, DMSO-d6 = 2.50 ppm). The following abbreviations were used in reporting spectra, br s (broad singlet), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets). Mass spectra were obtained on Agilent LC-ESI-MS system. All analytical HPLC were carried out on Agilent system. Water with 0.1% TFA and acetonitrile with 0.1% TFA were used as eluents and the flow rate was 0.5 mL/min. -

What Are Their Roles in Mitochondrial Protein Synthesis?
Characterisation of human mtRF1 and C12orf65: What are their roles in mitochondrial protein synthesis? Aleksandra Pajak M.Res Thesis submitted to Newcastle University in candidature for the degree of Doctor of Philosophy Newcastle University Faculty of Medical Sciences Institute for Ageing and Health Mitochondrial Research Group January 2013 Abstract Mitochondria have their own protein synthesis machinery that synthesises the oxidative phosphorylation components encoded by their mtDNA. This translation process consists of four main phases: initiation, elongation, termination and ribosome recycling. Termination and its control have been the least investigated. Recently, however, the termination factor, mtRF1a, has been characterised as sufficient to release all the nascent proteins from the mitoribosome. Furthermore, bioinformatics has identified three additional members of this mitochondrial release factor family namely, mtRF1, C12orf65 and ICT1. The latter is now known to be incorporated into the mitoribosome but its exact function remains unclear. My project has therefore focussed on characterising the remaining two factors; mtRF1 and C12orf65, and investigating their possible involvement in mitochondrial protein synthesis. It has been demonstrated that protein synthesis is not perfect and bacterial ribosomes not infrequently stall during translation. This can result from limiting amounts of charged tRNAs, stable secondary structures, or truncated/degraded transcripts. Ribosome stalling has been shown to cause growth arrest. In order to prevent that and maintain high efficiency of mitochondrial protein synthesis such stalled complexes need to be rapidly recycled. Bacteria have developed at least three distinct mechanisms by which ribosomes can be rescued. Contrastingly, despite the presence of truncated mRNAs in mitochondria, no such quality control mechanisms have been identified in these organelles. -

Reprogramming of Trna Modifications Controls the Oxidative Stress Response by Codon-Biased Translation of Proteins
Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Chan, Clement T.Y. et al. “Reprogramming of tRNA Modifications Controls the Oxidative Stress Response by Codon-biased Translation of Proteins.” Nature Communications 3 (2012): 937. As Published http://dx.doi.org/10.1038/ncomms1938 Publisher Nature Publishing Group Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/76775 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins Clement T.Y. Chan,1,2 Yan Ling Joy Pang,1 Wenjun Deng,1 I. Ramesh Babu,1 Madhu Dyavaiah,3 Thomas J. Begley3 and Peter C. Dedon1,4* 1Department of Biological Engineering, 2Department of Chemistry and 4Center for Environmental Health Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139; 3College of Nanoscale Science and Engineering, University at Albany, SUNY, Albany, NY 12203 * Corresponding author: PCD, Department of Biological Engineering, NE47-277, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139; tel 617-253-8017; fax 617-324-7554; email [email protected] 2 ABSTRACT Selective translation of survival proteins is an important facet of cellular stress response. We recently demonstrated that this translational control involves a stress-specific reprogramming of modified ribonucleosides in tRNA. -

Protein Name Accession Number Molecular Weight Myovi-GTD
MyoVI-GTD MyoVI-GTD MyoVa-MGT MyoVa-MGT Molecular Spectral Unique Spectral Unique Protein Name Accession Number Weight Counts Peptides Counts Peptides Dync1h1 Cytoplasmic dynein 1 heavy chain 1 IPI00119876 532 kDa 310 121 515 182 Spna2 Spectrin alpha 2 IPI00757353 285 kDa 853 170 597 149 Myo5a 215 kDa protein IPI00875222 215 kDa 162 47 874 109 AU042671 hypothetical protein LOC269700 isoform 1 IPI00762814 453 kDa 2 2 231 104 Spnb2 Isoform 1 of Spectrin beta chain, brain 1 IPI00319830 274 kDa 505 122 347 100 Dmxl2 Isoform 1 of DmX-like protein 2 IPI00853932 338 kDa 63 38 251 100 Cltc Clathrin heavy chain 1 IPI00169916 (+1) 192 kDa 1994 138 565 90 Mtap2 12 days embryo spinal cord cDNA, RIKEN full-length enriched library, clone:C530026F16 product:microtubule-associated protein 2, full insert sequenceIPI00894724 199 kDa 229 82 258 74 Mtap1a Isoform 1 of Microtubule-associated protein 1A IPI00408909 (+1) 300 kDa 310 86 214 74 Itpr1 Isoform 4 of Inositol 1,4,5-trisphosphate receptor type 1 IPI00230019 (+3) 311 kDa 37 18 155 73 Huwe1 HECT, UBA and WWE domain containing 1 IPI00463909 (+1) 483 kDa 5 5 91 69 Fasn Fatty acid synthase IPI00113223 272 kDa 24 17 140 68 Usp9x Ubiquitin carboxyl-terminal hydrolase IPI00798468 291 kDa 68 45 98 65 Lrp1 Prolow-density lipoprotein receptor-related protein 1 precursor IPI00119063 505 kDa 92 53 109 62 Myh10 Myosin-10 IPI00515398 (+1) 229 kDa 65 40 98 59 Mical1 NEDD9-interacting protein with calponin homology and LIM domains IPI00116371 117 kDa 2 2 203 57 Plec1 Isoform PLEC-1I of Plectin-1 IPI00229509 (+10) -

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

Analysis of the Relationship Between Ribosomal Protein and SSU Processome Assembly in Saccharomyces Cerevisiae
Analysis of the relationship between ribosomal protein and SSU processome assembly in Saccharomyces cerevisiae Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) der naturwissenschaftlichen Fakultät III – Biologie und vorklinische Medizin - der Universität Regensburg vorgelegt von Steffen Jakob aus Wolfen Januar 2010 Promotionsgesuch eingereicht am: 13. Januar 2010 Die Arbeit wurde angeleitet von: Prof. Dr. Herbert Tschochner Prüfungsausschuss: Vorsitzender: Prof. Dr. Armin Kurtz 1. Prüfer: Prof. Dr. Herbert Tschochner 2. Prüfer: Prof. Dr. Rainer Deutzmann 3. Prüfer: Prof. Dr. Wolfgang Seufert Tag der mündlichen Prüfung: 24. März 2010 Die vorliegende Arbeit wurde in der Zeit von April 2006 bis Januar 2010 am Lehrstuhl Biochemie III des Institutes für Biochemie, Genetik und Mikrobiologie der Naturwissenschaftlichen Fakultät III der Universität zu Regensburg unter Anleitung von Dr. Philipp Milkereit im Labor von Prof. Dr. Herbert Tschochner angefertigt. Ich erkläre hiermit, dass ich diese Arbeit selbst verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel verwendet habe. Diese Arbeit war bisher noch nicht Bestandteil eines Prüfungsverfahrens. Andere Promotionsversuche wurden nicht unternommen. Regensburg, den 13. Januar 2010 Steffen Jakob Table of Contents Table of Contents 1 SUMMARY ...................................................................................................... 1 2 INTRODUCTION ............................................................................................ -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -
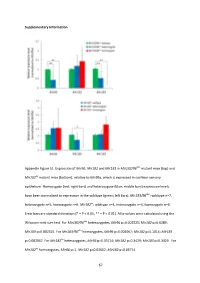
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Micrornas Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins
cells Review microRNAs Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins Abu Musa Md Talimur Reza 1,2 and Yu-Guo Yuan 1,3,* 1 Jiangsu Co-Innovation Center of Prevention and Control of Important Animal Infectious Diseases and Zoonoses, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China; [email protected] 2 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawi´nskiego5a, 02-106 Warsaw, Poland 3 Jiangsu Key Laboratory of Zoonosis/Joint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China * Correspondence: [email protected]; Tel.: +86-514-8797-9228 Abstract: Ribosomal proteins (RPs) are mostly derived from the energy-consuming enzyme families such as ATP-dependent RNA helicases, AAA-ATPases, GTPases and kinases, and are important structural components of the ribosome, which is a supramolecular ribonucleoprotein complex, composed of Ribosomal RNA (rRNA) and RPs, coordinates the translation and synthesis of proteins with the help of transfer RNA (tRNA) and other factors. Not all RPs are indispensable; in other words, the ribosome could be functional and could continue the translation of proteins instead of lacking in some of the RPs. However, the lack of many RPs could result in severe defects in the biogenesis of ribosomes, which could directly influence the overall translation processes and global expression of the proteins leading to the emergence of different diseases including cancer. While microRNAs (miRNAs) are small non-coding RNAs and one of the potent regulators of the post-transcriptional 0 gene expression, miRNAs regulate gene expression by targeting the 3 untranslated region and/or coding region of the messenger RNAs (mRNAs), and by interacting with the 50 untranslated region, Citation: Reza, A.M.M.T.; Yuan, Y.-G. -
Attachment PDF Icon
Spectrum Name of Protein Count of Peptides Ratio (POL2RA/IgG control) POLR2A_228kdBand POLR2A DNA-directed RNA polymerase II subunit RPB1 197 NOT IN CONTROL IP POLR2A_228kdBand POLR2B DNA-directed RNA polymerase II subunit RPB2 146 NOT IN CONTROL IP POLR2A_228kdBand RPAP2 Isoform 1 of RNA polymerase II-associated protein 2 24 NOT IN CONTROL IP POLR2A_228kdBand POLR2G DNA-directed RNA polymerase II subunit RPB7 23 NOT IN CONTROL IP POLR2A_228kdBand POLR2H DNA-directed RNA polymerases I, II, and III subunit RPABC3 19 NOT IN CONTROL IP POLR2A_228kdBand POLR2C DNA-directed RNA polymerase II subunit RPB3 17 NOT IN CONTROL IP POLR2A_228kdBand POLR2J RPB11a protein 7 NOT IN CONTROL IP POLR2A_228kdBand POLR2E DNA-directed RNA polymerases I, II, and III subunit RPABC1 8 NOT IN CONTROL IP POLR2A_228kdBand POLR2I DNA-directed RNA polymerase II subunit RPB9 9 NOT IN CONTROL IP POLR2A_228kdBand ALMS1 ALMS1 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2D DNA-directed RNA polymerase II subunit RPB4 6 NOT IN CONTROL IP POLR2A_228kdBand GRINL1A;Gcom1 Isoform 12 of Protein GRINL1A 6 NOT IN CONTROL IP POLR2A_228kdBand RECQL5 Isoform Beta of ATP-dependent DNA helicase Q5 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2L DNA-directed RNA polymerases I, II, and III subunit RPABC5 5 NOT IN CONTROL IP POLR2A_228kdBand KRT6A Keratin, type II cytoskeletal 6A 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2K DNA-directed RNA polymerases I, II, and III subunit RPABC4 2 NOT IN CONTROL IP POLR2A_228kdBand RFC4 Replication factor C subunit 4 1 NOT IN CONTROL IP POLR2A_228kdBand RFC2