Propeller Protein Containing a FYVE Domain, Prof: an Adaptor for Kinases Involved in Glucose Uptake in Adipocytes
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Structure and Function of the Fgd Family of Divergent FYVE Domain Proteins
Biochemistry and Cell Biology Structure and Function of the Fgd Family of Divergent FYVE Domain Proteins Journal: Biochemistry and Cell Biology Manuscript ID bcb-2018-0185.R1 Manuscript Type: Mini Review Date Submitted by the 03-Aug-2018 Author: Complete List of Authors: Eitzen, Gary; University of Alberta Faculty of Medicine and Dentistry Smithers, Cameron C.; University of Alberta, Biochemistry Murray, Allan; University of Alberta Faculty of Medicine and Dentistry Overduin, Michael; University of Alberta Faculty of Medicine and Dentistry Draft Fgd, Pleckstrin Homology domain, FYVE domain, Dbl Homology Domain, Keyword: Rho GEF Is the invited manuscript for consideration in a Special CSMB Special Issue Issue? : https://mc06.manuscriptcentral.com/bcb-pubs Page 1 of 37 Biochemistry and Cell Biology Title: Structure and Function of the Fgd Family of Divergent FYVE Domain Proteins Authors: Gary Eitzen1, Cameron C. Smithers2, Allan G Murray3 and Michael Overduin2* Draft 1Department of Cell Biology, 2Department of Biochemistry, 3Department of Medicine, University of Alberta, Edmonton, Alberta, Canada *Corresponding author. Michael Overduin Telephone: +1 780 492 3518 Fax: +1 780 492-0886 E-mail: [email protected] https://mc06.manuscriptcentral.com/bcb-pubs Biochemistry and Cell Biology Page 2 of 37 Abstract FYVE domains are highly conserved protein modules that typically bind phosphatidylinositol 3-phosphate (PI3P) on the surface of early endosomes. Along with pleckstrin homology (PH) and phox homology (PX) domains, FYVE domains are the principal readers of the phosphoinositide (PI) code that mediate specific recognition of eukaryotic organelles. Of all the human FYVE domain-containing proteins, those within the Faciogenital dysplasia (Fgd) subfamily are particularly divergent, and couple with GTPases to exert unique cellular functions. -

Molecular Mechanism of Membrane Targeting by the GRP1 PH Domain
Supplemental Material can be found at: http://www.jlr.org/cgi/content/full/M800150-JLR200/DC1 Molecular mechanism of membrane targeting by the GRP1 PH domain † † † Ju He,* Rachel M. Haney, ,§ Mohsin Vora, Vladislav V. Verkhusha,** Robert V. Stahelin, ,§ and Tatiana G. Kutateladze1,* Department of Pharmacology,* University of Colorado Health Sciences Center, Aurora, CO; † Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, South Bend, IN; Department of Chemistry and Biochemistry and The Walther Center for Cancer Research,§ University of Notre Dame, South Bend, IN; and Department of Anatomy and Structural Biology,** Downloaded from Albert Einstein College of Medicine, Bronx, NY Abstract The general receptor for phosphoinositides iso- Supplementary key words general receptor for phosphoinositides iso- • • • form 1 (GRP1) is recruited to the plasma membrane in re- form 1 pleckstrin homology domain phosphoinositide phosphati- dylinositol 3,4,5-trisphosphate sponse to activation of phosphoinositide 3-kinases and www.jlr.org accumulation of phosphatidylinositol 3,4,5-trisphosphate ʼ [PtdIns(3,4,5)P3]. GRP1 s pleckstrin homology (PH) do- main recognizes PtdIns(3,4,5)P3 with high specificity and af- The signaling lipid phosphatidylinositol 3,4,5-trisphos- finity, however, the precise mechanism of its association phate [PtdIns(3,4,5)P3] is produced in plasma membranes at Albert Einstein College of Medicine Library on July 14, 2008 with membranes remains unclear. Here, we detail the mo- in response to stimulation of cell surface receptors by lecular basis of membrane anchoring by the GRP1 PH do- growth factors and hormones (1). Class I phosphoinositide main. Our data reveal a multivalent membrane docking (PI) 3-kinases phosphorylate the inositol headgroup of the involving PtdIns(3,4,5)P binding, regulated by pH and fa- 3 relatively abundant phosphatidylinositol 4,5-bisphosphate cilitated by electrostatic interactions with other anionic lip- [Ptdns(4,5)P2], transiently elevating the level of PtdIns ids. -

The PX Domain Protein Interaction Network in Yeast
The PX domain protein interaction network in yeast Zur Erlangung des akademischen Grades eines DOKTORS DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät für Chemie und Biowissenschaften der Universität Karlsruhe (TH) vorgelegte DISSERTATION von Dipl. Biol. Carolina S. Müller aus Buenos Aires Dekan: Prof. Dr. Manfred Kappes Referent: Dr. Nils Johnsson Korreferent: HD. Dr. Adam Bertl Tag der mündlichen Prüfung: 17.02.2005 I dedicate this work to my Parents and Alex TABLE OF CONTENTS Table of contents Introduction 1 Yeast as a model organism in proteome analysis 1 Protein-protein interactions 2 Protein Domains in Yeast 3 Classification of protein interaction domains 3 Phosphoinositides 5 Function 5 Structure 5 Biochemistry 6 Localization 7 Lipid Binding Domains 8 The PX domain 10 Function of PX domain containing proteins 10 PX domain structure and PI binding affinities 10 Yeast PX domain containing proteins 13 PX domain and protein-protein interactions 13 Lipid binding domains and protein-protein interactions 14 The PX-only proteins Grd19p and Ypt35p and their phenotypes 15 Aim of my PhD work 16 Project outline 16 Searching for interacting partners 16 Confirmation of obtained interactions via a 16 second independent method Mapping the interacting region 16 The Two-Hybrid System 17 Definition 17 Basic Principle of the classical Yeast-Two Hybrid System 17 Peptide Synthesis 18 SPOT synthesis technique 18 Analysis of protein- peptide contact sites based on SPOT synthesis 19 TABLE OF CONTENTS Experimental procedures 21 Yeast two-hybrid assay -

Membrane Insertion of the FYVE Domain Is Modulated by Ph Ju He,1 Mohsin Vora,2 Rachel M
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Membrane insertion of the FYVE domain is modulated by pH Ju He,1 Mohsin Vora,2 Rachel M. Haney,2,3 Grigory S. Filonov,4 Catherine A. Musselman,1 Christopher G. Burd,5 Andrei G. Kutateladze,6 Vladislav V. Verkhusha,4 Robert V. Stahelin,2,3,7* and Tatiana G. Kutateladze1* 1 Department of Pharmacology, University of Colorado Denver School of Medicine, Aurora, Colorado 80045 2 Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, South Bend, Indiana 46617 3 Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556 4 Department of Anatomy and Structural Biology, Albert Einstein College of Medicine, Bronx, New York 10461 5 Department of Cell and Developmental Biology, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 19104 6 Department of Chemistry and Biochemistry, University of Denver, Denver, Colorado 80210 7 The Walther Center for Cancer Research, University of Notre Dame, Notre Dame, Indiana 46556 INTRODUCTION ABSTRACT Phosphoinositide (PI) 3-kinases regulate membrane trafficking, pro- The FYVE domain associates with phosphati- tein sorting and signaling by generating phosphatidylinositol (PtdIns) dylinositol 3-phosphate [PtdIns(3)P] in mem- derivatives phosphorylated at the third position of the inositol ring.1,2 branes of early endosomes and penetrates bilayers. Here, we detail principles of mem- Of the four known products of PI 3-kinases, PtdIns 3-phosphate brane anchoring and show that the FYVE do- [PtdIns(3)P] is the most abundant and is constitutively produced in the main insertion into PtdIns(3)P-enriched cytosolic leaflet of membranes of early endosomes. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

Solving the FYVE Domain–Ptdins(3)P Puzzle
© 2001 Nature Publishing Group http://structbio.nature.com news and views Solving the FYVE domain–PtdIns(3)P puzzle Paul C. Driscoll Recent crystallographic analyses of membrane-tethering FYVE finger domains from proteins involved in the regulation of endocytic vesicle trafficking have led to conflicting views of the precise nature of the contacts formed with the specific phospholipid ligand. New NMR data obtained for ligand-bound forms of a FYVE domain help resolve the atomic details of this interaction. The truly dynamic nature of eukaryotic cell membranes is brought into sharp focus by the text book description of receptor-mediated endocytosis: ∼50% of the plasma membrane is internalized and recycled every hour whereas the synthesis of new membrane is perhaps one tenth of this rate. The highly regulated process of endocytosis, by which cells recover fluid, chemicals and specific macromolecules from the external environment, is the tar- get of intense investigation. The interplay of cytosolic proteins with constituent plasma and endosomal membranes pro- vides many challenges to structural biolo- gists, not least at the interface between the soluble components and the membrane lipids themselves. Progress by the tradi- tional methods of structural investigation at this ‘phase boundary’ is particularly difficult. © http://structbio.nature.com Group 2001 Nature Publishing In a paper published recently in Science, Kutateladze and Overduin1 report an extension of their earlier work using NMR spectroscopy to analyze the lipid interactions of the FYVE protein domain from the protein early endosome Fig. 1 The chemical structure of PtdIns(3)P and the predicted ‘side-on’ interaction with the FYVE antigen-1 (EEA1). -

Cellular Functions of Phosphatidylinositol 3-Phosphate and FYVE Domain Proteins David J
Biochem. J. (2001) 355, 249–258 (Printed in Great Britain) 249 REVIEW ARTICLE Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins David J. GILLOOLY, Anne SIMONSEN and Harald STENMARK1 Department of Biochemistry, The Norwegian Radium Hospital, Montebello, 0310 Oslo, Norway PtdIns3P is a phosphoinositide 3-kinase product that has been identified which contain a FYVE domain, and in this review we strongly implicated in regulating membrane trafficking in both discuss the functions of PtdIns3P and its FYVE-domain-con- mammalian and yeast cells. PtdIns3P has been shown to be taining effector proteins in membrane trafficking, cytoskeletal specifically located on membranes associated with the endocytic regulation and receptor signalling. pathway. Proteins that contain FYVE zinc-finger domains are recruited to PtdIns3P-containing membranes. Structural in- formation is now available concerning the interaction between Key words: membrane traffic, phosphoinositide, phospho- FYVE domains and PtdIns3P. A number of proteins have been inositide 3-kinase, protein domains, recruitment. INTRODUCTION PtdIns(3,4,5)P$. PtdIns(3,4,5)P$ is produced upon agonist stimu- Phosphorylated derivatives of PtdIns, known as phospho- lation in mammalian cells and interacts with a number of inositides, play a key role in the membrane recruitment and\or proteins containing pleckstrin homology (PH) domains [3,5]. activation of proteins [1]. In this way they provide a means for Cellular PtdIns(3,4)P# is probably mainly produced from a the temporal and spatial regulation of complex cellular processes. dephosphorylation of PtdIns(3,4,5)P$, since the kinetics of its The products of phosphoinositide 3-kinases (PI 3-kinases) production often immediately follow on from a transient have been the subject of much investigation and have been PtdIns(3,4,5)P$ production [6]. -

Supplemental Table 3 Two-Class Paired Significance Analysis of Microarrays Comparing Gene Expression Between Paired
Supplemental Table 3 Two‐class paired Significance Analysis of Microarrays comparing gene expression between paired pre‐ and post‐transplant kidneys biopsies (N=8). Entrez Fold q‐value Probe Set ID Gene Symbol Unigene Name Score Gene ID Difference (%) Probe sets higher expressed in post‐transplant biopsies in paired analysis (N=1871) 218870_at 55843 ARHGAP15 Rho GTPase activating protein 15 7,01 3,99 0,00 205304_s_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 6,30 4,50 0,00 1563649_at ‐‐ ‐‐ ‐‐ 6,24 3,51 0,00 1567913_at 541466 CT45‐1 cancer/testis antigen CT45‐1 5,90 4,21 0,00 203932_at 3109 HLA‐DMB major histocompatibility complex, class II, DM beta 5,83 3,20 0,00 204606_at 6366 CCL21 chemokine (C‐C motif) ligand 21 5,82 10,42 0,00 205898_at 1524 CX3CR1 chemokine (C‐X3‐C motif) receptor 1 5,74 8,50 0,00 205303_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 5,68 6,87 0,00 226841_at 219972 MPEG1 macrophage expressed gene 1 5,59 3,76 0,00 203923_s_at 1536 CYBB cytochrome b‐245, beta polypeptide (chronic granulomatous disease) 5,58 4,70 0,00 210135_s_at 6474 SHOX2 short stature homeobox 2 5,53 5,58 0,00 1562642_at ‐‐ ‐‐ ‐‐ 5,42 5,03 0,00 242605_at 1634 DCN decorin 5,23 3,92 0,00 228750_at ‐‐ ‐‐ ‐‐ 5,21 7,22 0,00 collagen, type III, alpha 1 (Ehlers‐Danlos syndrome type IV, autosomal 201852_x_at 1281 COL3A1 dominant) 5,10 8,46 0,00 3493///3 IGHA1///IGHA immunoglobulin heavy constant alpha 1///immunoglobulin heavy 217022_s_at 494 2 constant alpha 2 (A2m marker) 5,07 9,53 0,00 1 202311_s_at -
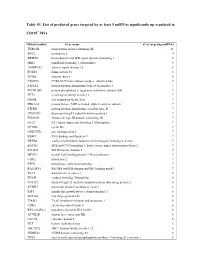
Table S3. List of Predicted Genes Targeted by at Least 5 Mirnas Significantly up Regulated In
Table S3. List of predicted genes targeted by at least 5 miRNAs significantly up regulated in CD105+ MVs. Official symbol Gene name n° of targeting miRNAs TNRC6B trinucleotide repeat containing 6B 11 BNC2 basonuclin 2 9 BRWD1 bromodomain and WD repeat domain containing 1 8 MIB1 mindbomb homolog 1 (Drosophila) 8 ANKRD52 ankyrin repeat domain 52 8 FOXP1 forkhead box P1 8 ITGB8 integrin, beta 8 8 CNOT6L CCR4-NOT transcription complex, subunit 6-like 8 PTP4A1 protein tyrosine phosphatase type IVA, member 1 7 PPP1R15B protein phosphatase 1, regulatory (inhibitor) subunit 15B 7 TET3 tet oncogene family member 3 7 GMFB glia maturation factor, beta 7 PRKAA2 protein kinase, AMP-activated, alpha 2 catalytic subunit 7 PTPRD protein tyrosine phosphatase, receptor type, D 7 TP53INP1 tumor protein p53 inducible nuclear protein 1 7 FNDC3B fibronectin type III domain containing 3B 7 FAT3 FAT tumor suppressor homolog 3 (Drosophila) 7 CCND2 cyclin D2 7 ONECUT2 one cut homeobox 2 7 RBM9 RNA binding motif protein 9 7 ERBB4 v-erb-a erythroblastic leukemia viral oncogene homolog 4 (avian) 7 BACH2 BTB and CNC homology 1, basic leucine zipper transcription factor 2 7 SMAD4 SMAD family member 4 7 MECP2 methyl CpG binding protein 2 (Rett syndrome) 7 UBN2 ubinuclein 2 7 PTEN phosphatase and tensin homolog 7 RALGPS1 Ral GEF with PH domain and SH3 binding motif 1 7 DLC1 deleted in liver cancer 1 6 ENAH enabled homolog (Drosophila) 6 NUFIP2 nuclear fragile X mental retardation protein interacting protein 2 6 KCMF1 potassium channel modulatory factor 1 6 IGF1 insulin-like growth factor 1 (somatomedin C) 6 ZNF148 zinc finger protein 148 6 TIAM1 T-cell lymphoma invasion and metastasis 1 6 CDK6 cyclin-dependent kinase 6 6 RP5-1022P6.2 hypothetical protein KIAA1434 6 ACVR2B activin A receptor, type IIB 6 CLCN5 chloride channel 5 6 HLF hepatic leukemia factor 6 TBC1D12 TBC1 domain family, member 12 6 FRMD4A FERM domain containing 4A 6 NUS1 nuclear undecaprenyl pyrophosphate synthase 1 homolog (S. -

Suppl Figure 1
Suppl Table 2. Gene Annotation (October 2011) for the selected genes used in the study. Locus Identifier Gene Model Description AT5G51780 basic helix-loop-helix (bHLH) DNA-binding superfamily protein; FUNCTIONS IN: DNA binding, sequence-specific DNA binding transcription factor activity; INVOLVED IN: regulation of transcription; LOCATED IN: nucleus; CONTAINS InterPro DOMAIN/s: Helix-loop-helix DNA-binding domain (InterPro:IPR001092), Helix-loop-helix DNA-binding (InterPro:IPR011598); BEST Arabidopsis thaliana protein match is: basic helix-loop-helix (bHLH) D AT3G53400 BEST Arabidopsis thaliana protein match is: conserved peptide upstream open reading frame 47 (TAIR:AT5G03190.1); Has 285 Blast hits to 285 proteins in 23 species: Archae - 0; Bacteria - 0; Metazoa - 1; Fungi - 0; Plants - 279; Viruses - 0; Other Eukaryotes - 5 (source: NCBI BLink). AT1G44760 Adenine nucleotide alpha hydrolases-like superfamily protein; FUNCTIONS IN: molecular_function unknown; INVOLVED IN: response to stress; EXPRESSED IN: 22 plant structures; EXPRESSED DURING: 13 growth stages; CONTAINS InterPro DOMAIN/s: UspA (InterPro:IPR006016), Rossmann-like alpha/beta/alpha sandwich fold (InterPro:IPR014729); BEST Arabidopsis thaliana protein match is: Adenine nucleotide alpha hydrolases-li AT4G19950 unknown protein; BEST Arabidopsis thaliana protein match is: unknown protein (TAIR:AT5G44860.1); Has 338 Blast hits to 330 proteins in 72 species: Archae - 2; Bacteria - 94; Metazoa - 7; Fungi - 0; Plants - 232; Viruses - 0; Other Eukaryotes - 3 (source: NCBI BLink). AT3G14280 -

Multipurpose Cellular Lipids with Emerging Roles in Cell Death
Cell Death & Differentiation (2019) 26:781–793 https://doi.org/10.1038/s41418-018-0269-2 REVIEW ARTICLE Phosphoinositides: multipurpose cellular lipids with emerging roles in cell death 1 1 1 1 1 1 Thanh Kha Phan ● Scott A Williams ● Guneet K Bindra ● Fung T Lay ● Ivan K. H Poon ● Mark D Hulett Received: 5 September 2018 / Revised: 18 December 2018 / Accepted: 19 December 2018 / Published online: 11 February 2019 © ADMC Associazione Differenziamento e Morte Cellulare 2019 Abstract Phosphorylated phosphatidylinositol lipids, or phosphoinositides, critically regulate diverse cellular processes, including signalling transduction, cytoskeletal reorganisation, membrane dynamics and cellular trafficking. However, phosphoinosi- tides have been inadequately investigated in the context of cell death, where they are mainly regarded as signalling secondary messengers. However, recent studies have begun to highlight the importance of phosphoinositides in facilitating cell death execution. Here, we cover the latest phosphoinositide research with a particular focus on phosphoinositides in the mechanisms of cell death. This progress article also raises key questions regarding the poorly defined role of phosphoinositides, particularly during membrane-associated events in cell death such as apoptosis and secondary necrosis. fi 1234567890();,: 1234567890();,: The review then further discusses important future directions for the phosphoinositide eld, including therapeutically targeting phosphoinositides to modulate cell death. Facts peptide-induced necrosis,