Structural Studies of Human Telomerase Complexes by Cryo‑Electron Microscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Female Fellows of the Royal Society
Female Fellows of the Royal Society Professor Jan Anderson FRS [1996] Professor Ruth Lynden-Bell FRS [2006] Professor Judith Armitage FRS [2013] Dr Mary Lyon FRS [1973] Professor Frances Ashcroft FMedSci FRS [1999] Professor Georgina Mace CBE FRS [2002] Professor Gillian Bates FMedSci FRS [2007] Professor Trudy Mackay FRS [2006] Professor Jean Beggs CBE FRS [1998] Professor Enid MacRobbie FRS [1991] Dame Jocelyn Bell Burnell DBE FRS [2003] Dr Philippa Marrack FMedSci FRS [1997] Dame Valerie Beral DBE FMedSci FRS [2006] Professor Dusa McDuff FRS [1994] Dr Mariann Bienz FMedSci FRS [2003] Professor Angela McLean FRS [2009] Professor Elizabeth Blackburn AC FRS [1992] Professor Anne Mills FMedSci FRS [2013] Professor Andrea Brand FMedSci FRS [2010] Professor Brenda Milner CC FRS [1979] Professor Eleanor Burbidge FRS [1964] Dr Anne O'Garra FMedSci FRS [2008] Professor Eleanor Campbell FRS [2010] Dame Bridget Ogilvie AC DBE FMedSci FRS [2003] Professor Doreen Cantrell FMedSci FRS [2011] Baroness Onora O'Neill * CBE FBA FMedSci FRS [2007] Professor Lorna Casselton CBE FRS [1999] Dame Linda Partridge DBE FMedSci FRS [1996] Professor Deborah Charlesworth FRS [2005] Dr Barbara Pearse FRS [1988] Professor Jennifer Clack FRS [2009] Professor Fiona Powrie FRS [2011] Professor Nicola Clayton FRS [2010] Professor Susan Rees FRS [2002] Professor Suzanne Cory AC FRS [1992] Professor Daniela Rhodes FRS [2007] Dame Kay Davies DBE FMedSci FRS [2003] Professor Elizabeth Robertson FRS [2003] Professor Caroline Dean OBE FRS [2004] Dame Carol Robinson DBE FMedSci -

Driving Diffusion of Scientific Innovation
Driving Diffusion of Scientific Innovation - The Role of Institutional Entrepreneurship and Open Science in Synthetic Biology Vom Promotionsausschuss der Technischen Universit¨at Hamburg-Harburg zur Erlangung des akademischen Grades Doktor der Wirtschafts- und Sozialwissenschaften (Dr. rer. pol.) genehmigte Dissertation von Giulio Barth aus M¨unster 2018 Advisors: Prof. Dr. C. Ihl, Prof. Dr. M. G. M¨ohrle Institute of Entrepreneurship, TUHH i Gutachter: Prof. Dr. C. Ihl Prof. Dr. M. G. Mohrle¨ Vorsitz: Prof. Dr. C. Luthje¨ Tag der mundlichen¨ Prufung:¨ 14. September 2018 ”Our victory: inevitable; our timing: uncertain.” (Drew Endy, Assistant Professor Stanford University) i Abstract Scientific innovations need to widely diffuse to fully exploit their potential. Prior research investigated levers on the diffusion of scientific innovation with particular interest on institutions, e.g., settings of property rights. As institutional theory lacks in explaining emergence and shaping of institu- tions, the institutional entrepreneur approach faces these limitations. Key actors combine logics from multiple fields and convince their social context of their ideas to legitimate the creation of new institutions and shape an emerging field. This thesis validates theories on institutional entrepreneurs and investigates the end-to process from diffusing a logic to the impact of an established insti- tution on scientific innovations in context of the emerging synthetic biology. The field is expected to introduce the 5th revolution and characterized by the central logic of making biology an engineering discipline. In chapter 4 theories on institutional entrepreneurs driving diffusion of in- stitutional logics to shape an emerging field are validated. To measure the social influence mechanisms, the heterogeneous diffusion model is adapted to the institutional logic. -

Mechanisms Directing Receptor-Specific Gene Regulation
Mechanisms Directing Receptor-Specific Gene Regulation by the Androgen and Glucocorticoid Receptor Inaugural-Dissertation to obtain the academic degree Doctor rerum naturalium (Dr. rer. nat.) submitted to the Department of Biology, Chemistry, Pharmacy of Freie Universität Berlin by Marina Kulik • 2021 The dissertation was prepared under the supervision of Dr. Sebastiaan H. Meijsing at the Max Planck Institute for Molecular Genetics in Berlin from September 2015 to February 2021. 1st Reviewer: Dr. Sebastiaan Meijsing 2nd Reviewer: Prof. Dr. Markus Wahl Date of defense: 21.05.2021 1 Selbstständigkeitserklärung Hiermit bestätige ich, dass ich die vorliegende Arbeit selbstständig und unter Zuhilfenahme der angegebenen Literatur erstellt habe. Acknowledgments First of all, I would like to express my gratitude to my supervisor Sebastiaan Meijsing for his support and guidance during my PhD. I would like to thank Martin Vingron for the opportunity to be part of his research group and for the great collaborations. Especially, I would like to thank Stefan Haas for introducing me to the world of RNA-seq and Gözde Kibar, who contributed with her bioinformatical analyses to this work. I would like to thank Sarah Kinkley for her support and for giving me the opportunity to finish my PhD in her group. I wish to thank Stefan Prekovic, Isabel Mayayo-Peralta and Wilbert Zwart from the NKI in Amsterdam for sharing their expertise in “nuclear signaling” and the great collaboration. I am particularly grateful to Melissa Bothe, her computational analyses and support in the lab contributed a lot to this work. My special gratitude goes to Laura Glaser for her continuous advice and for freezing my cells in the evening countless times. -

EMBO Facts & Figures
excellence in life sciences Reykjavik Helsinki Oslo Stockholm Tallinn EMBO facts & figures & EMBO facts Copenhagen Dublin Amsterdam Berlin Warsaw London Brussels Prague Luxembourg Paris Vienna Bratislava Budapest Bern Ljubljana Zagreb Rome Madrid Ankara Lisbon Athens Jerusalem EMBO facts & figures HIGHLIGHTS CONTACT EMBO & EMBC EMBO Long-Term Fellowships Five Advanced Fellows are selected (page ). Long-Term and Short-Term Fellowships are awarded. The Fellows’ EMBO Young Investigators Meeting is held in Heidelberg in June . EMBO Installation Grants New EMBO Members & EMBO elects new members (page ), selects Young EMBO Women in Science Young Investigators Investigators (page ) and eight Installation Grantees Gerlind Wallon EMBO Scientific Publications (page ). Programme Manager Bernd Pulverer S Maria Leptin Deputy Director Head A EMBO Science Policy Issues report on quotas in academia to assure gender balance. R EMBO Director + + A Conducts workshops on emerging biotechnologies and on H T cognitive genomics. Gives invited talks at US National Academy E IC of Sciences, International Summit on Human Genome Editing, I H 5 D MAN 201 O N Washington, DC.; World Congress on Research Integrity, Rio de A M Janeiro; International Scienti c Advisory Board for the Centre for Eilish Craddock IT 2 015 Mammalian Synthetic Biology, Edinburgh. Personal Assistant to EMBO Fellowships EMBO Scientific Publications EMBO Gold Medal Sarah Teichmann and Ido Amit receive the EMBO Gold the EMBO Director David del Álamo Thomas Lemberger Medal (page ). + Programme Manager Deputy Head EMBO Global Activities India and Singapore sign agreements to become EMBC Associate + + Member States. EMBO Courses & Workshops More than , participants from countries attend 6th scienti c events (page ); participants attend EMBO Laboratory Management Courses (page ); rst online course EMBO Courses & Workshops recorded in collaboration with iBiology. -

Smutty Alchemy
University of Calgary PRISM: University of Calgary's Digital Repository Graduate Studies The Vault: Electronic Theses and Dissertations 2021-01-18 Smutty Alchemy Smith, Mallory E. Land Smith, M. E. L. (2021). Smutty Alchemy (Unpublished doctoral thesis). University of Calgary, Calgary, AB. http://hdl.handle.net/1880/113019 doctoral thesis University of Calgary graduate students retain copyright ownership and moral rights for their thesis. You may use this material in any way that is permitted by the Copyright Act or through licensing that has been assigned to the document. For uses that are not allowable under copyright legislation or licensing, you are required to seek permission. Downloaded from PRISM: https://prism.ucalgary.ca UNIVERSITY OF CALGARY Smutty Alchemy by Mallory E. Land Smith A THESIS SUBMITTED TO THE FACULTY OF GRADUATE STUDIES IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY GRADUATE PROGRAM IN ENGLISH CALGARY, ALBERTA JANUARY, 2021 © Mallory E. Land Smith 2021 MELS ii Abstract Sina Queyras, in the essay “Lyric Conceptualism: A Manifesto in Progress,” describes the Lyric Conceptualist as a poet capable of recognizing the effects of disparate movements and employing a variety of lyric, conceptual, and language poetry techniques to continue to innovate in poetry without dismissing the work of other schools of poetic thought. Queyras sees the lyric conceptualist as an artistic curator who collects, modifies, selects, synthesizes, and adapts, to create verse that is both conceptual and accessible, using relevant materials and techniques from the past and present. This dissertation responds to Queyras’s idea with a collection of original poems in the lyric conceptualist mode, supported by a critical exegesis of that work. -

Nucleosome Repeat Length and Linker Histone Stoichiometry Determine Chromatin Fiber Structure
Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure Andrew Routh, Sara Sandin, and Daniela Rhodes† Laboratory of Molecular Biology, Medical Research Council, Hills Road, Cambridge CB2 0QH, United Kingdom Edited by Alan R. Fersht, University of Cambridge, Cambridge, United Kingdom, and approved April 7, 2008 (received for review March 10, 2008) To understand how nuclear processes involving DNA are regulated, 11). The linker histone, on binding to the nucleosome core, not knowledge of the determinants of chromatin condensation is only constrains an additional 20 bp of DNA (12, 13), but also required. From recent structural studies it has been concluded that determines the trajectory of the DNA entering and exiting the the formation of the 30-nm chromatin fiber does not require the nucleosome (14, 15), which in turn will direct the relative linker histone. Here, by comparing the linker histone-dependent positioning of successive nucleosomes in an array. Consequently, compaction of long, reconstituted nucleosome arrays with differ- the linker histone is likely to have a pivotal role in the formation ent nucleosome repeat lengths (NRLs), 167 and 197 bp, we of chromatin higher-order structures (16). Besides a structural establish that the compaction behavior is both NRL- and linker role, it is also emerging that the linker histone and its subtypes histone-dependent. Only the 197-bp NRL array can form 30-nm have diverse biologically important roles (reviewed in refs. 3 and higher-order chromatin structure. Importantly for understanding 17). A gene knockout of three of the six murine H1 gene variants the regulation of compaction, this array shows a cooperative linker is embryonic lethal (18), whereas single knockouts modulate histone-dependent compaction. -
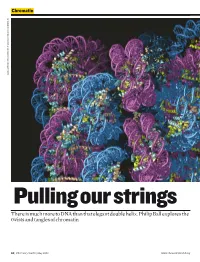
There Is Much More to DNA Than That Elegant Double Helix. Philip Ball Explores the Twists and Tangles of Chromatin
Chromatin DANIELA RHODES / MRC LABORATORY OF MOLECULAR BIOLOGY, CAMBRIDGE, UK CAMBRIDGE, BIOLOGY, MOLECULAR OF LABORATORY MRC / RHODES DANIELA Pulling our strings There is much more to DNA than that elegant double helix. Philip Ball explores the twists and tangles of chromatin 50 | Chemistry World | May 2008 www.chemistryworld.org Why isn’t my arm a leg, or my liver read and transcribed into RNA a kidney? Their cells all have the In short – the first step in the conversion same genes. But genetics isn’t just Chromosomes are of genetic information to proteins. about what genes you have – it also made up of a fibrous To understand why some genes depends on how you use them. composite of DNA and are silent while others are actively This crucial aspect of our biological protein called chromatin transcribed – and thus what allows identities has been overshadowed by This coiled network genetically identical cells to become a focus on the identity of our genes, of protein fibres plays differentiated into different tissue and how these differ from those of an important role in types – we need to unravel the other people and other species. But it controlling the activity of packaging. has now become clear that genetics an organism’s genes can’t be fully understood until we Chromatin can be Wound up genes have a better picture, not just of what changed and remodelled In eukaryotic cells, where the is in our genes, but of how they are with chemical labels DNA is housed in a cell nucleus, labelled and packaged. that provide signals for the chromosomes aren’t pure This evolving view of gene enzymes, indicating DNA. -

New Chief Executive As MRC Gets Cash Boost
AUTUMN 2007 News from the Medical Research Council New Chief Executive as MRC gets cash boost A new Chief Executive, Sir Leszek Borysiewicz, took the helm of the MRC at the beginning of October. Sir Leszek, who comes from Imperial College London where he was Deputy Rector, spoke of his excitement about leading the MRC at a time of change and opportunity for the organisation: “I’m thrilled by the chance to work across the whole spectrum of biomedical science and to help to make a difference in relation to healthcare for individuals in the UK and globally.” The chairman of the MRC, Sir John Chisholm, welcomed Sir Leszek: “He is the perfect person to lead the MRC in the new environment of coordinated health research in the UK. I am delighted he’ll join us – his stature as a scientist and clinician reflects the importance of the role the MRC will play in a coordinated strategy for turning research findings into healthcare.” Timely arrival Sir Leszek joined the MRC just days before the Chancellor announced a significant increase in the organisation’s budget, from £543 million to £682 million a year by 2010. In his pre-budget report to the House of Commons on 10 October, Alistair Darling explained that he was funding recognised across the world. And so more British medical discovery can in full the recommendations of Sir David Cooksey’s review of publicly be translated into new health drugs, treatments and preventions… We funded health research including a single strategy for health research in will expand the single fund for health research to £1.7 billion by 2010.” the UK overseen by an Office for the Strategic Coordination of Health Research (OSCHR). -

Analysis of Genome Instability Using Genomic and Bioinformatic Approaches
FACULTY OF SCIENCE Analysis of genome instability using genomic and bioinformatic approaches Ph.D. Thesis Kateřina Havlová Supervisor: prof. RNDr. Jiří Fajkus, CSc. Laboratory of Functional Genomics and Proteomics Brno 2020 Bibliographic Entry Author: Mgr. Kateřina Havlová Faculty of Science, Masaryk University National Centre for Biomolecular Research Functional Genomics and Proteomics Central European Institute of Technology, MU Mendel Centre for Plant Genomics and Proteomics Analysis of genome instability using genomic and Title of Thesis: bioinformatic approaches Degree programme: Genomics and proteomics Supervisor: prof. RNDr. Jiří Fajkus, CSc. Academic Year: 2019/2020 Number of Pages: 80+33 Keywords: rDNA; ribosomal DNA; ribosomal DNA intergenic spacer; chromatin; Arabidopsis thaliana; Chromatin Assembly factor 1; Physcomitrella patens; G-quadruplex Bibliografický záznam Autor: Mgr. Kateřina Havlová Přírodovědecká fakulta, Masarykova univerzita Národní centrum pro výzkum biomolekul Funkční genomika a proteomika Středoevropský technologický institut, MU Mendelovo centrum genomiky a proteomiky rostlin Analýza nestability genomu pomocí genomických a Název práce: bioinformatických přístupů Studijní program: Genomika a proteomika Vedoucí práce: prof. RNDr. Jiří Fajkus, CSc. Akademický rok: 2019/2020 Počet stran: 80+33 Klíčová slova: rDNA; ribosomální DNA; mezerník ribosomální DNA; chromatin; Arabidopsis thaliana; Chromatin Assembly factor 1; Physcomitrella patens; G-kvadruplex Abstract Ribosomal RNA genes (rDNA) are the most abundant and utilized genes in eukaryotes. They compose a vast portion of the genome and they are involved in the maintenance of the genome-wide chromatin structure. This thesis focuses on rDNA in two different model species – Arabidopsis thaliana and Physcomitrella patens. Firstly, we present here the detailed characterisation of the variability in the sequence of Arabidopsis intergenic spacer (IGS), the regulatory region separating each two copies of rRNA genes. -
Phd Thesis Antonina Zappone.Pdf
Abstract ABSTRACT In vertebrates, the UUAGGG telomere repeat containing long non-coding RNA TERRA is prone to form RNA:DNA hybrids at telomeres resulting the formation of R-loop structures, replicative stress and telomere instability. RNA:DNA hybrids represent a threat to genomic stability, but also contribute to recombination based alternative lengthening of telomeres (ALT). My PhD thesis aimed to identifie novel TERRA interactors involved in regulation of RNA:DNA hybrids (project 1) and to obtain new insights into the molecular function of TERRA interactrors in telomere regulation by identifying novel proteins (project 2). Here, we identify the TERRA binding proteins SFPQ as novel regulators of RNA:DNA hybrid related telomere instability. NONO and SFPQ locate at telomeres and have a common role in suppressing RNA:DNA hybrids and replication defects at telomeres. SFPQ acts as a barrier for homologous recombination at telomeres, thereby impacting on telomere length homeostasis. Our data identifies the RNA binding proteins SFPQ as novel regulators at telomeres that collaborate to ensure telomere integrity by suppressing telomere fragility and homologous recombination triggered by RNA:DNA hybrids. Because of lack of enzymatic activity in SFPQ, we performed a mass spectrometry analysis and we found DAXX as novel SFPQ interacting protein. After the identification od SFPQ interacting domain with DAXX, we carried out preliminary experiment to evaluate the role od DAXX in DNA damage activation, in induction of replication defects and telomere -

Feamle Fellows 2015
Female Fellows of the Royal Society Fellowship Professor Jan M Anderson FRS [1996] Dame Julia Higgins DBE FREng FRS [1995] Professor Judith Armitage FRS [2013] Professor Brigid Hogan FRS [2001] Professor Frances Ashcroft FMedSci FRS [1999] Professor Christine Holt FMedSci FRS [2009] Professor Gillian Bates FMedSci FRS [2007] Professor Judith Howard CBE FRS [2002] Professor Jean Beggs CBE FRS [1998] Professor Patricia Jacobs OBE FMedSci FRS [1993] Dame Jocelyn Bell Burnell DBE FRS [2003] Professor Lisa Jardine CBE FRS [2015]* Dame Valerie Beral DBE FMedSci FRS [2006] Dame Carole Jordan DBE FRS [1990] Dr Mariann Bienz FMedSci FRS [2003] Professor Victoria Kaspi FRS [2010] Professor Dorothy Bishop FBA FMedSci FRS [2014] Dr Olga Kennard OBE FRS [1987] Professor Elizabeth Blackburn AC FRS [1992] Professor Frances Kirwan DBE FRS [2001] Professor Andrea Brand FMedSci FRS [2010] Professor Jane Langdale FRS [2015] Professor Eleanor Burbidge FRS [1964] Professor Ottoline Leyser CBE FRS [2007] Professor Eleanor Campbell FRS [2010] Professor Ruth Lynden-Bell FRS [2006] Professor Doreen Cantrell CBE FMedSci FRS [2011] Professor Georgina Mace CBE FRS [2002] Professor Deborah Charlesworth FRS [2005] Professor Trudy Mackay FRS [2006] Professor Jennifer Clack FRS [2009] Professor Enid MacRobbie FRS [1991] Professor Jane Clarke FMedSci FRS [2015] Dr Philippa Marrack FMedSci FRS [1997] Professor Nicola Clayton FRS [2010] Professor Dusa McDuff FRS [1994] Professor Suzanne Cory AC FRS [1992] Professor Angela McLean FRS [2009] Professor Anne Cutler FRS [2015] -

Encountering the Organization
issue 14 winter 2009 | 2010 promoting excellence in the molecular life sciences in europe Encountering the organization During the past 10 years researchers and their groups in member states indeed lives up to its original promise: to iden- EMBO has awarded around where research infrastructures are, despite all tify and foster talented scientists and to dis- 2,000 post-doctoral fellow- efforts, still insuffi ciently developed. seminate ideas, knowledge and technology ships, elected about 200 More than 5,000 scientists participate across borders. It does so in a particularly Young Investigators and sup- every year in the 70 to 80 events supported via sensible way by generating and sustaining its ported over 1800 researchers EMBO Courses & Workshops Programme. This communities and by catalysing interactions via short-term fellowships. Moreover, since the traditional core programme is now comple- between them (see fi gure 1). start of the respective programme in 2006, mented by our annual broad-scope conference Having all these fi ne programmes up and 34 young group leaders received support The EMBO Meeting and by the EMBO|EMBL running, can we lean back in satisfaction? Of through EMBO Installation Grants. The unques- Symposia. course not! There are considerable challenges tioned quality of these programmes, which This year’s inaugural life science confer- ahead of us. address the gifted individual researcher, is ence – The EMBO Meeting – in Amsterdam Taking, for example, a closer look at our based as well on the selection process in which was exciting and spirited. It attracted over activities throughout our member states we can rely on the expertise of our dedicated 1,300 participants, and I am confi dent that it reveals that even though EMBO is engaged membership, as we rely on EMBO policy to will develop into a major annual event in life in all of them, there are asymmetries, which offer opportunities for its awardees to interact, sciences in Europe.