Mechanisms Directing Receptor-Specific Gene Regulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Female Fellows of the Royal Society
Female Fellows of the Royal Society Professor Jan Anderson FRS [1996] Professor Ruth Lynden-Bell FRS [2006] Professor Judith Armitage FRS [2013] Dr Mary Lyon FRS [1973] Professor Frances Ashcroft FMedSci FRS [1999] Professor Georgina Mace CBE FRS [2002] Professor Gillian Bates FMedSci FRS [2007] Professor Trudy Mackay FRS [2006] Professor Jean Beggs CBE FRS [1998] Professor Enid MacRobbie FRS [1991] Dame Jocelyn Bell Burnell DBE FRS [2003] Dr Philippa Marrack FMedSci FRS [1997] Dame Valerie Beral DBE FMedSci FRS [2006] Professor Dusa McDuff FRS [1994] Dr Mariann Bienz FMedSci FRS [2003] Professor Angela McLean FRS [2009] Professor Elizabeth Blackburn AC FRS [1992] Professor Anne Mills FMedSci FRS [2013] Professor Andrea Brand FMedSci FRS [2010] Professor Brenda Milner CC FRS [1979] Professor Eleanor Burbidge FRS [1964] Dr Anne O'Garra FMedSci FRS [2008] Professor Eleanor Campbell FRS [2010] Dame Bridget Ogilvie AC DBE FMedSci FRS [2003] Professor Doreen Cantrell FMedSci FRS [2011] Baroness Onora O'Neill * CBE FBA FMedSci FRS [2007] Professor Lorna Casselton CBE FRS [1999] Dame Linda Partridge DBE FMedSci FRS [1996] Professor Deborah Charlesworth FRS [2005] Dr Barbara Pearse FRS [1988] Professor Jennifer Clack FRS [2009] Professor Fiona Powrie FRS [2011] Professor Nicola Clayton FRS [2010] Professor Susan Rees FRS [2002] Professor Suzanne Cory AC FRS [1992] Professor Daniela Rhodes FRS [2007] Dame Kay Davies DBE FMedSci FRS [2003] Professor Elizabeth Robertson FRS [2003] Professor Caroline Dean OBE FRS [2004] Dame Carol Robinson DBE FMedSci -

Driving Diffusion of Scientific Innovation
Driving Diffusion of Scientific Innovation - The Role of Institutional Entrepreneurship and Open Science in Synthetic Biology Vom Promotionsausschuss der Technischen Universit¨at Hamburg-Harburg zur Erlangung des akademischen Grades Doktor der Wirtschafts- und Sozialwissenschaften (Dr. rer. pol.) genehmigte Dissertation von Giulio Barth aus M¨unster 2018 Advisors: Prof. Dr. C. Ihl, Prof. Dr. M. G. M¨ohrle Institute of Entrepreneurship, TUHH i Gutachter: Prof. Dr. C. Ihl Prof. Dr. M. G. Mohrle¨ Vorsitz: Prof. Dr. C. Luthje¨ Tag der mundlichen¨ Prufung:¨ 14. September 2018 ”Our victory: inevitable; our timing: uncertain.” (Drew Endy, Assistant Professor Stanford University) i Abstract Scientific innovations need to widely diffuse to fully exploit their potential. Prior research investigated levers on the diffusion of scientific innovation with particular interest on institutions, e.g., settings of property rights. As institutional theory lacks in explaining emergence and shaping of institu- tions, the institutional entrepreneur approach faces these limitations. Key actors combine logics from multiple fields and convince their social context of their ideas to legitimate the creation of new institutions and shape an emerging field. This thesis validates theories on institutional entrepreneurs and investigates the end-to process from diffusing a logic to the impact of an established insti- tution on scientific innovations in context of the emerging synthetic biology. The field is expected to introduce the 5th revolution and characterized by the central logic of making biology an engineering discipline. In chapter 4 theories on institutional entrepreneurs driving diffusion of in- stitutional logics to shape an emerging field are validated. To measure the social influence mechanisms, the heterogeneous diffusion model is adapted to the institutional logic. -

Androgen Receptor Interacting Proteins and Coregulators Table
ANDROGEN RECEPTOR INTERACTING PROTEINS AND COREGULATORS TABLE Compiled by: Lenore K. Beitel, Ph.D. Lady Davis Institute for Medical Research 3755 Cote Ste Catherine Rd, Montreal, Quebec H3T 1E2 Canada Telephone: 514-340-8260 Fax: 514-340-7502 E-Mail: [email protected] Internet: http://androgendb.mcgill.ca Date of this version: 2010-08-03 (includes articles published as of 2009-12-31) Table Legend: Gene: Official symbol with hyperlink to NCBI Entrez Gene entry Protein: Protein name Preferred Name: NCBI Entrez Gene preferred name and alternate names Function: General protein function, categorized as in Heemers HV and Tindall DJ. Endocrine Reviews 28: 778-808, 2007. Coregulator: CoA, coactivator; coR, corepressor; -, not reported/no effect Interactn: Type of interaction. Direct, interacts directly with androgen receptor (AR); indirect, indirect interaction; -, not reported Domain: Interacts with specified AR domain. FL-AR, full-length AR; NTD, N-terminal domain; DBD, DNA-binding domain; h, hinge; LBD, ligand-binding domain; C-term, C-terminal; -, not reported References: Selected references with hyperlink to PubMed abstract. Note: Due to space limitations, all references for each AR-interacting protein/coregulator could not be cited. The reader is advised to consult PubMed for additional references. Also known as: Alternate gene names Gene Protein Preferred Name Function Coregulator Interactn Domain References Also known as AATF AATF/Che-1 apoptosis cell cycle coA direct FL-AR Leister P et al. Signal Transduction 3:17-25, 2003 DED; CHE1; antagonizing regulator Burgdorf S et al. J Biol Chem 279:17524-17534, 2004 CHE-1; AATF transcription factor ACTB actin, beta actin, cytoplasmic 1; cytoskeletal coA - - Ting HJ et al. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

EMBO Facts & Figures
excellence in life sciences Reykjavik Helsinki Oslo Stockholm Tallinn EMBO facts & figures & EMBO facts Copenhagen Dublin Amsterdam Berlin Warsaw London Brussels Prague Luxembourg Paris Vienna Bratislava Budapest Bern Ljubljana Zagreb Rome Madrid Ankara Lisbon Athens Jerusalem EMBO facts & figures HIGHLIGHTS CONTACT EMBO & EMBC EMBO Long-Term Fellowships Five Advanced Fellows are selected (page ). Long-Term and Short-Term Fellowships are awarded. The Fellows’ EMBO Young Investigators Meeting is held in Heidelberg in June . EMBO Installation Grants New EMBO Members & EMBO elects new members (page ), selects Young EMBO Women in Science Young Investigators Investigators (page ) and eight Installation Grantees Gerlind Wallon EMBO Scientific Publications (page ). Programme Manager Bernd Pulverer S Maria Leptin Deputy Director Head A EMBO Science Policy Issues report on quotas in academia to assure gender balance. R EMBO Director + + A Conducts workshops on emerging biotechnologies and on H T cognitive genomics. Gives invited talks at US National Academy E IC of Sciences, International Summit on Human Genome Editing, I H 5 D MAN 201 O N Washington, DC.; World Congress on Research Integrity, Rio de A M Janeiro; International Scienti c Advisory Board for the Centre for Eilish Craddock IT 2 015 Mammalian Synthetic Biology, Edinburgh. Personal Assistant to EMBO Fellowships EMBO Scientific Publications EMBO Gold Medal Sarah Teichmann and Ido Amit receive the EMBO Gold the EMBO Director David del Álamo Thomas Lemberger Medal (page ). + Programme Manager Deputy Head EMBO Global Activities India and Singapore sign agreements to become EMBC Associate + + Member States. EMBO Courses & Workshops More than , participants from countries attend 6th scienti c events (page ); participants attend EMBO Laboratory Management Courses (page ); rst online course EMBO Courses & Workshops recorded in collaboration with iBiology. -

Smutty Alchemy
University of Calgary PRISM: University of Calgary's Digital Repository Graduate Studies The Vault: Electronic Theses and Dissertations 2021-01-18 Smutty Alchemy Smith, Mallory E. Land Smith, M. E. L. (2021). Smutty Alchemy (Unpublished doctoral thesis). University of Calgary, Calgary, AB. http://hdl.handle.net/1880/113019 doctoral thesis University of Calgary graduate students retain copyright ownership and moral rights for their thesis. You may use this material in any way that is permitted by the Copyright Act or through licensing that has been assigned to the document. For uses that are not allowable under copyright legislation or licensing, you are required to seek permission. Downloaded from PRISM: https://prism.ucalgary.ca UNIVERSITY OF CALGARY Smutty Alchemy by Mallory E. Land Smith A THESIS SUBMITTED TO THE FACULTY OF GRADUATE STUDIES IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY GRADUATE PROGRAM IN ENGLISH CALGARY, ALBERTA JANUARY, 2021 © Mallory E. Land Smith 2021 MELS ii Abstract Sina Queyras, in the essay “Lyric Conceptualism: A Manifesto in Progress,” describes the Lyric Conceptualist as a poet capable of recognizing the effects of disparate movements and employing a variety of lyric, conceptual, and language poetry techniques to continue to innovate in poetry without dismissing the work of other schools of poetic thought. Queyras sees the lyric conceptualist as an artistic curator who collects, modifies, selects, synthesizes, and adapts, to create verse that is both conceptual and accessible, using relevant materials and techniques from the past and present. This dissertation responds to Queyras’s idea with a collection of original poems in the lyric conceptualist mode, supported by a critical exegesis of that work. -

New Approach for Untangling the Role of Uncommon Calcium-Binding Proteins in the Central Nervous System
brain sciences Review New Approach for Untangling the Role of Uncommon Calcium-Binding Proteins in the Central Nervous System Krisztina Kelemen * and Tibor Szilágyi Department of Physiology, Doctoral School, Faculty of Medicine, George Emil Palade University of Medicine, Pharmacy, Science, and Technology of Targu Mures, 540142 Târgu Mures, , Romania; [email protected] * Correspondence: [email protected]; Tel.: +40-746-248064 Abstract: Although Ca2+ ion plays an essential role in cellular physiology, calcium-binding proteins (CaBPs) were long used for mainly as immunohistochemical markers of specific cell types in different regions of the central nervous system. They are a heterogeneous and wide-ranging group of proteins. Their function was studied intensively in the last two decades and a tremendous amount of informa- tion was gathered about them. Girard et al. compiled a comprehensive list of the gene-expression profiles of the entire EF-hand gene superfamily in the murine brain. We selected from this database those CaBPs which are related to information processing and/or neuronal signalling, have a Ca2+- buffer activity, Ca2+-sensor activity, modulator of Ca2+-channel activity, or a yet unknown function. In this way we created a gene function-based selection of the CaBPs. We cross-referenced these findings with publicly available, high-quality RNA-sequencing and in situ hybridization databases (Human Protein Atlas (HPA), Brain RNA-seq database and Allen Brain Atlas integrated into the HPA) and created gene expression heat maps of the regional and cell type-specific expression levels of the selected CaBPs. This represents a useful tool to predict and investigate different expression patterns and functions of the less-known CaBPs of the central nervous system. -

Human Park7/DJ-1 Antibody Catalog Number: ATGA0292
Human Park7/DJ-1 antibody Catalog Number: ATGA0292 PRODUCT INPORMATION Catalog number ATGA0292 Clone No. AT1E12 Product type Monoclonal Antibody UnitProt No. Q99497 NCBI Accession No. NP_009193 Alternative Names Parkinson disease protein 7, PARK7, DJ-1 (PARK7), Oncogene DJ1, Parkinson disease protein 7, Protein DJ-1, Parkinson disease protein 7 CAP1, DJ1, DJ1 protein, Oncogene DJ1, SP22 PRODUCT SPECIFICATION Antibody Host Mouse Reacts With Human Concentration 1mg/ml (determined by BCA assay) Formulation Liquid in. Phosphate-Buffered Saline (pH 7.4) with 0.02% Sodium Azide, 10% glycerol Immunogen Recombinant human Park7/DJ-1 (1-189aa) purified from E. coli Isotype IgG2b kappa Purification Note By protein-A affinity chromatography Application ELISA,WB,ICC/IF,FACS Usage The antibody has been tested by ELISA, Western blot, ICC/IF and FACS analysis to assure specificity and reactivity. Since application varies, however, each investigation should be titrated by the reagent to obtain optimal results. 1 Human Park7/DJ-1 antibody Catalog Number: ATGA0292 Storage Can be stored at +2C to +8C for 1 week. For long term storage, aliquot and store at -20C to -80C. Avoid repeated freezing and thawing cycles. BACKGROUND Description Parkinson disease (autosomal recessive, early onset) 7, also known as PARK7/DJ-1, has been shown to interact with EFCAB6 and protein inhibitor of activated STAT2. Defects in PARK7 are the cause of autosomal recessive early-onset Parkinson's disease 7. This protein belongs to the peptidase C56 family of proteins. It acts as a positive regulator of androgen receptor-dependent transcription. It may also function as a redox-sensitive chaperone, as a sensor for oxidative stress, and it apparently protects neurons against oxidative stress and cell death. -

Novel Mutations Segregating with Complete Androgen Insensitivity Syndrome and Their Molecular Characteristics
International Journal of Molecular Sciences Article Novel Mutations Segregating with Complete Androgen Insensitivity Syndrome and Their Molecular Characteristics Agnieszka Malcher 1, Piotr Jedrzejczak 2, Tomasz Stokowy 3 , Soroosh Monem 1, Karolina Nowicka-Bauer 1, Agnieszka Zimna 1, Adam Czyzyk 4, Marzena Maciejewska-Jeske 4, Blazej Meczekalski 4, Katarzyna Bednarek-Rajewska 5, Aldona Wozniak 5, Natalia Rozwadowska 1 and Maciej Kurpisz 1,* 1 Institute of Human Genetics, Polish Academy of Sciences, 60-479 Poznan, Poland; [email protected] (A.M.); [email protected] (S.M.); [email protected] (K.N.-B.); [email protected] (A.Z.); [email protected] (N.R.) 2 Division of Infertility and Reproductive Endocrinology, Department of Gynecology, Obstetrics and Gynecological Oncology, Poznan University of Medical Sciences, 60-535 Poznan, Poland; [email protected] 3 Department of Clinical Science, University of Bergen, 5020 Bergen, Norway; [email protected] 4 Department of Gynecological Endocrinology, Poznan University of Medical Sciences, 60-535 Poznan, Poland; [email protected] (A.C.); [email protected] (M.M.-J.); [email protected] (B.M.) 5 Department of Clinical Pathology, Poznan University of Medical Sciences, 60-355 Poznan, Poland; [email protected] (K.B.-R.); [email protected] (A.W.) * Correspondence: [email protected]; Tel.: +48-61-6579-202 Received: 11 October 2019; Accepted: 29 October 2019; Published: 30 October 2019 Abstract: We analyzed three cases of Complete Androgen Insensitivity Syndrome (CAIS) and report three hitherto undisclosed causes of the disease. RNA-Seq, Real-timePCR, Western immunoblotting, and immunohistochemistry were performed with the aim of characterizing the disease-causing variants. -

Nucleosome Repeat Length and Linker Histone Stoichiometry Determine Chromatin Fiber Structure
Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure Andrew Routh, Sara Sandin, and Daniela Rhodes† Laboratory of Molecular Biology, Medical Research Council, Hills Road, Cambridge CB2 0QH, United Kingdom Edited by Alan R. Fersht, University of Cambridge, Cambridge, United Kingdom, and approved April 7, 2008 (received for review March 10, 2008) To understand how nuclear processes involving DNA are regulated, 11). The linker histone, on binding to the nucleosome core, not knowledge of the determinants of chromatin condensation is only constrains an additional 20 bp of DNA (12, 13), but also required. From recent structural studies it has been concluded that determines the trajectory of the DNA entering and exiting the the formation of the 30-nm chromatin fiber does not require the nucleosome (14, 15), which in turn will direct the relative linker histone. Here, by comparing the linker histone-dependent positioning of successive nucleosomes in an array. Consequently, compaction of long, reconstituted nucleosome arrays with differ- the linker histone is likely to have a pivotal role in the formation ent nucleosome repeat lengths (NRLs), 167 and 197 bp, we of chromatin higher-order structures (16). Besides a structural establish that the compaction behavior is both NRL- and linker role, it is also emerging that the linker histone and its subtypes histone-dependent. Only the 197-bp NRL array can form 30-nm have diverse biologically important roles (reviewed in refs. 3 and higher-order chromatin structure. Importantly for understanding 17). A gene knockout of three of the six murine H1 gene variants the regulation of compaction, this array shows a cooperative linker is embryonic lethal (18), whereas single knockouts modulate histone-dependent compaction. -

Translational Profiling Reveals the Transcriptome of Leptin Receptor Neurons and Its Regulation by Leptin
TRANSLATIONAL PROFILING REVEALS THE TRANSCRIPTOME OF LEPTIN RECEPTOR NEURONS AND ITS REGULATION BY LEPTIN by Margaret B. Allison A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Molecular and Integrative Physiology) In the University of Michigan 2015 Doctoral Committee: Professor Martin G. Myers Jr., Chair Associate Professor Carol F. Elias Professor Malcolm J. Low Professor Suzanne Moenter Professor Audrey Seasholtz Before you leave these portals To meet less fortunate mortals There's just one final message I would give to you: You all have learned reliance On the sacred teachings of science So I hope, through life, you never will decline In spite of philistine defiance To do what all good scientists do: Experiment! -- Cole Porter There is no cure for curiosity. -- unknown © Margaret Brewster Allison 2015 ACKNOWLEDGEMENTS If it takes a village to raise a child, it takes a research university to raise a graduate student. There are many people who have supported me over the past six years at Michigan, and it is hard to imagine pursuing my PhD without them. First and foremost among all the people I need to thank is my mentor, Martin. Nothing I might say here would ever suffice to cover the depth and breadth of my gratitude to him. Without his patience, his insight, and his at times insufferably positive outlook, I don’t know where I would be today. Martin supported my intellectual curiosity, honed my scientific inquiry, and allowed me to do some really fun research in his lab. It was a privilege and a pleasure to work for him and with him. -
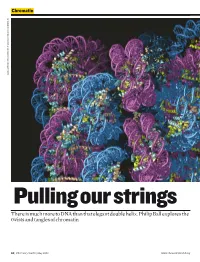
There Is Much More to DNA Than That Elegant Double Helix. Philip Ball Explores the Twists and Tangles of Chromatin
Chromatin DANIELA RHODES / MRC LABORATORY OF MOLECULAR BIOLOGY, CAMBRIDGE, UK CAMBRIDGE, BIOLOGY, MOLECULAR OF LABORATORY MRC / RHODES DANIELA Pulling our strings There is much more to DNA than that elegant double helix. Philip Ball explores the twists and tangles of chromatin 50 | Chemistry World | May 2008 www.chemistryworld.org Why isn’t my arm a leg, or my liver read and transcribed into RNA a kidney? Their cells all have the In short – the first step in the conversion same genes. But genetics isn’t just Chromosomes are of genetic information to proteins. about what genes you have – it also made up of a fibrous To understand why some genes depends on how you use them. composite of DNA and are silent while others are actively This crucial aspect of our biological protein called chromatin transcribed – and thus what allows identities has been overshadowed by This coiled network genetically identical cells to become a focus on the identity of our genes, of protein fibres plays differentiated into different tissue and how these differ from those of an important role in types – we need to unravel the other people and other species. But it controlling the activity of packaging. has now become clear that genetics an organism’s genes can’t be fully understood until we Chromatin can be Wound up genes have a better picture, not just of what changed and remodelled In eukaryotic cells, where the is in our genes, but of how they are with chemical labels DNA is housed in a cell nucleus, labelled and packaged. that provide signals for the chromosomes aren’t pure This evolving view of gene enzymes, indicating DNA.