The Role of Histone Modifications in Severe Asthma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cancer Immunity (1 December 2006) Vol
Cancer Immun 1424-9634Academy of Cancer Immunology Cancer Immunity (1 December 2006) Vol. 6, p. 12 Submitted: 26 September 2006. Accepted: 10 October 2006. Copyright © 2006 by Andrew J. G. Simpson 061012 Article Physical interaction of two cancer-testis antigens, MAGE-C1 (CT7) and NY-ESO-1 (CT6) Hearn J. Cho1*,**, Otavia L. Caballero2*, Sacha Gnjatic2, Valéria C. C. Andrade3, Gisele W. Colleoni3, Andre L. Vettore4, Hasina H. Outtz1, Sheila Fortunato2, Nasser Altorki1, Cathy A. Ferrera1, Ramon Chua2, Achim A. Jungbluth2, Yao-Tseng Chen1, Lloyd J. Old2 and Andrew J. G. Simpson2 1Weill Medical College of Cornell University, 1300 York Avenue, New York, NY 10021, USA 2Ludwig Institute for Cancer Research, New York Branch at Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021, USA 3Escola Paulista de Medicina, Universidade Federal de Sao Paulo, Sao Paulo, SP, Brazil 4Ludwig Institute for Cancer Research, Sao Paulo Branch, Sao Paulo, SP, Brazil *These authors contributed equally to this work **Present address: NYU Cancer Institute, New York University School of Medicine, 550 First Avenue, New York, NY 10016, USA Contributed by: LJ Old Cancer/testis (CT) antigens are the protein products of germ line- encoded on the X chromosome (CT-X antigens) and those that associated genes that are activated in a wide variety of tumors and can are not (non-X CT antigens) (1). elicit autologous cellular and humoral immune responses. CT It is estimated that 10% of the genes on the X-chromosome antigens can be divided between those that are encoded on the X belong to CT-X families (5). -

Food Allergy Outline
Allergy Evaluation-What it all Means & Role of Allergist Sai R. Nimmagadda, M.D.. Associated Allergists and Asthma Specialists Ltd. Clinical Assistant Professor Of Pediatrics Northwestern University Chicago, Illinois Objectives of Presentation • Discuss the different options for allergy evaluation. – Skin tests – Immunocap Testing • Understand the results of Allergy testing in various allergic diseases. • Briefly Understand what an Allergist Does Common Allergic Diseases Seen in the Primary Care Office • Atopic Dermatitis/Eczema • Food Allergy • Allergic Rhinitis • Allergic Asthma • Allergic GI Diseases Factors that Influence Allergies Development and Expression Host Factors Environmental Factors . Genetic Indoor allergens - Atopy Outdoor allergens - Airway hyper Occupational sensitizers responsiveness Tobacco smoke . Gender Air Pollution . Obesity Respiratory Infections Diet © Global Initiative for Asthma Why Perform Allergy Testing? – Confirm Allergens and answer specific questions. • Am I allergic to my dog? • Do I have a milk allergy? • Have I outgrown my allergy? • Do I need medications? • Am I penicillin allergic? • Do I have a bee sting allergy Tests Performed in the Diagnostic Allergy Laboratory • Allergen-specific IgE (over 200 allergen extracts) – Pollen (weeds, grasses, trees), – Epidermal, dust mites, molds, – Foods, – Venoms, – Drugs, – Occupational allergens (e.g., natural rubber latex) • Total Serum IgE (anti-IgE; ABPA) • Multi-allergen screen for IgE antibody Diagnostic Allergy Testing Serological Confirmation of Sensitization History of RAST Testing • RAST (radioallergosorbent test) invented and marketed in 1974 • The suspected allergen is bound to an insoluble material and the patient's serum is added • If the serum contains antibodies to the allergen, those antibodies will bind to the allergen • Radiolabeled anti-human IgE antibody is added where it binds to those IgE antibodies already bound to the insoluble material • The unbound anti-human IgE antibodies are washed away. -
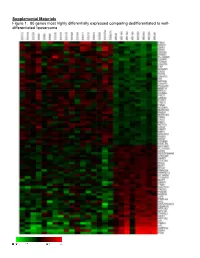
Supplemental Materials Figure 1. 80 Genes Most Highly Differentially Expressed Comparing Dedifferentiated to Well- Differentiated Liposarcoma
Supplemental Materials Figure 1. 80 genes most highly differentially expressed comparing dedifferentiated to well- differentiated liposarcoma Figure 2. Validation of microa CDC2, RACGAP1, FGFR-2, MAD2 and CITED1 200 CDK4 by Liposarcoma Subtype 16 0 12 0 80 Expression40 Level 0 Nl Fat rray data by quantitative RT-P Well Diff 16 0 p16 by Liposarcoma Subtype Dediff 12 0 80 Myxoid Expression40 Level Round Cell 0 12 0 MDM2 by Liposarcoma Subtype 10 0 Pleomorphic Nl Fat 80 60 40 Expression Level 20 RACGAP1 by LiposarcomaWell Diff Subtype 0 70 CR for genes CDK4, MDM2, p16, 60 Dediff 50 Nl Fat 40 30 Myxoid Expression20 Level Well Diff 10 50 0 Round Cell CDC2 by Liposarcoma Subtype Dediff 40 Nl Fat Pleomorphic 30 Myxoid 20 Expression Level Well Diff 10 MAD2L1 by Liposarcoma Subtype Round Cell 60 0 50 Dediff Pleomorphic 40 Nl Fat 30 Myxoid 20 Expression Level Well Diff 10 FGFR-2 by Liposarcoma Subtype 0 Round Cell 200 16 0 Dediff Nl Fat Pleomorphic 12 0 Myxoid 80 Expression Level Well Diff 40 Round Cell 0 Dediff Pleomorphic Nl Fat Myxoid Well Diff CITED1 by Liposarcoma Subtype Round Cell 10 0 80 Dediff Pleomorphic 60 40 Myxoid Expression Level 20 0 Round Cell Nl Fat Pleomorphic Well Diff Dediff Myxoid Round Cell Pleomorphic Table 1. 142 Gene classifier for liposarcoma subtype The genes used for each pair wise subtype comparison are grouped together. The flag column indicates which genes are unique to each subtype comparison. The values show the mean expression levels (actually the mean of the log expression levels was computed and than transformed back to absolute expression level). -

Expression of Cancer-Testis Antigens MAGEA1, MAGEA3, ACRBP, PRAME, SSX2, and CTAG2 in Myxoid and Round Cell Liposarcoma
Modern Pathology (2014) 27, 1238–1245 1238 & 2014 USCAP, Inc All rights reserved 0893-3952/14 $32.00 Expression of cancer-testis antigens MAGEA1, MAGEA3, ACRBP, PRAME, SSX2, and CTAG2 in myxoid and round cell liposarcoma Jessica A Hemminger1, Amanda Ewart Toland2, Thomas J Scharschmidt3, Joel L Mayerson3, Denis C Guttridge2 and O Hans Iwenofu1 1Department of Pathology and Laboratory Medicine, Wexner Medical Center at The Ohio State University, Columbus, OH, USA; 2Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University Wexner Medical Center, Columbus, OH, USA and 3Department of Orthopedics, The Ohio State University Wexner Medical Center, Columbus, OH, USA Myxoid and round-cell liposarcoma is a frequently encountered liposarcoma subtype. The mainstay of treatment remains surgical excision with or without chemoradiation. However, treatment options are limited in the setting of metastatic disease. Cancer-testis antigens are immunogenic antigens with the expression largely restricted to testicular germ cells and various malignancies, making them attractive targets for cancer immunotherapy. Gene expression studies have reported the expression of various cancer-testis antigens in liposarcoma, with mRNA expression of CTAG1B, CTAG2, MAGEA9, and PRAME described specifically in myxoid and round-cell liposarcoma. Herein, we further explore the expression of the cancer-testis antigens MAGEA1, ACRBP, PRAME, and SSX2 in myxoid and round-cell liposarcoma by immunohistochemistry in addition to determining mRNA levels of CTAG2 (LAGE-1), PRAME, and MAGEA3 by quantitative real-time PCR. Samples in formalin-fixed paraffin-embedded blocks (n ¼ 37) and frozen tissue (n ¼ 8) were obtained for immunohistochemistry and quantitative real-time PCR, respectively. Full sections were stained with antibodies to MAGEA1, ACRBP, PRAME, and SSX2 and staining was assessed for intensity (1–2 þ ) and percent tumor positivity. -
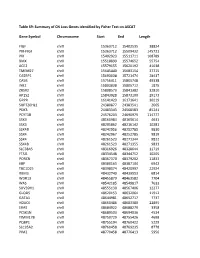
Table S9: Summary of CN Loss Genes Identified by Fisher Test on ASCAT
Table S9: Summary of CN Loss Genes identified by Fisher Test on ASCAT Gene Symbol Chromosome Start End Length FIGF chrX 15363712 15402535 38824 PIR-FIGF chrX 15363712 15509432 145721 PIR chrX 15402923 15511711 108789 BMX chrX 15518899 15574652 55754 ACE2 chrX 15579155 15620192 41038 TMEM27 chrX 15645440 15683154 37715 CA5BP1 chrX 15693038 15721474 28437 CA5B chrX 15756411 15805748 49338 INE2 chrX 15803838 15805712 1875 ZRSR2 chrX 15808573 15841382 32810 AP1S2 chrX 15843928 15873100 29173 GRPR chrX 16141423 16171641 30219 SUPT20HL1 chrX 24380877 24383541 2665 PDK3 chrX 24483343 24568583 85241 PCYT1B chrX 24576203 24690979 114777 SSX9 chrX 48160984 48165614 4631 SSX3 chrX 48205862 48216142 10281 SSX4B chrX 48242956 48252785 9830 SSX4 chrX 48242967 48252785 9819 SSX4 chrX 48261523 48271344 9822 SSX4B chrX 48261523 48271355 9833 SLC38A5 chrX 48316926 48328644 11719 FTSJ1 chrX 48334548 48344752 10205 PORCN chrX 48367370 48379202 11833 EBP chrX 48380163 48387104 6942 TBC1D25 chrX 48398074 48420997 22924 RBM3 chrX 48432740 48439553 6814 WDR13 chrX 48455879 48463582 7704 WAS chrX 48542185 48549817 7633 SUV39H1 chrX 48555130 48567406 12277 GLOD5 chrX 48620153 48632064 11912 GATA1 chrX 48644981 48652717 7737 HDAC6 chrX 48660486 48683380 22895 ERAS chrX 48684922 48688279 3358 PCSK1N chrX 48689503 48694036 4534 TIMM17B chrX 48750729 48755426 4698 PQBP1 chrX 48755194 48760422 5229 SLC35A2 chrX 48760458 48769235 8778 PIM2 chrX 48770458 48776413 5956 OTUD5 chrX 48779302 48815648 36347 KCND1 chrX 48818638 48828251 9614 GRIPAP1 chrX 48830133 48858675 28543 -

RAST Type Tests CPT: 86003
Medicare Local Coverage Determination Policy Allergy Testing RAST Type Tests CPT: 86003 Medically Supportive CMS Policy for Kentucky and Ohio ICD Codes are listed Local policies are determined by the performing test location. This is determined by the state on subsequent page(s) in which your performing laboratory resides and where your testing is commonly performed. of this document. Coverage Indications, Limitations, and/or Medical Necessity Radioallergosorbent test (RAST), fluoroallergosorbent test (FAST), and multiple antigen simultaneous tests are in vitro techniques for Add full policy information determining whether a patient's serum contains IgE antibodies against specific allergens of clinical importance. As with any allergy testing, the need for such tests is based on the findings during a complete history and physical examination of the patient. The multiple antigen simultaneous testing technique is similar to the RAST/FAST techniques in that it depends upon the existence of Template structure: allergic antibodies in the blood of the patient being tested. With the multiple antigen simultaneous test system, several antigens may be used to test for specific IgE simultaneously. First level is for headers such as limitations, ELISA (enzyme-linked immunosorbent assay) is another in vitro method of allergy testing for specific IgE antibodies against allergens. indications and usage guidelines This method is also a variation of RAST. Second level is for main body copy Limitations • Third level is for bullet (if needed) The following tests are considered to be not medically necessary and will be denied. • ELISA/Act qualitative antibody testing. This testing is used to determine in vitro reaction to various foods and relies on lymphocyte blastogenesis in response to certain food antigens. -

NY-ESO-1 (CTAG1B) Expression in Mesenchymal Tumors
Modern Pathology (2015) 28, 587–595 & 2015 USCAP, Inc. All rights reserved 0893-3952/15 $32.00 587 NY-ESO-1 (CTAG1B) expression in mesenchymal tumors Makoto Endo1,2,7, Marieke A de Graaff3,7, Davis R Ingram4, Simin Lim1, Dina C Lev4, Inge H Briaire-de Bruijn3, Neeta Somaiah5, Judith VMG Bove´e3, Alexander J Lazar6 and Torsten O Nielsen1 1Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, British Columbia, Canada; 2Department of Orthopaedic Surgery, Kyushu University, Fukuoka, Japan; 3Department of Pathology, Leiden University Medical Center, Leiden, The Netherlands; 4Department of Surgical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 5Department of Sarcoma Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA and 6Department of Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA New York esophageal squamous cell carcinoma 1 (NY-ESO-1, CTAG1B) is a cancer-testis antigen and currently a focus of several targeted immunotherapeutic strategies. We performed a large-scale immunohistochemical expression study of NY-ESO-1 using tissue microarrays of mesenchymal tumors from three institutions in an international collaboration. A total of 1132 intermediate and malignant and 175 benign mesenchymal lesions were enrolled in this study. Immunohistochemical staining was performed on tissue microarrays using a monoclonal antibody for NY-ESO-1. Among mesenchymal tumors, myxoid liposarcomas showed the highest positivity for NY-ESO-1 (88%), followed by synovial sarcomas (49%), myxofibrosarcomas (35%), and conventional chondrosarcomas (28%). Positivity of NY-ESO-1 in the remaining mesenchymal tumors was consistently low, and no immunoreactivity was observed in benign mesenchymal lesions. -

Gene Expression Profiling Using Nanostring Digital RNA Counting to Identify Potential Target Antigens for Melanoma Immunotherapy
Published OnlineFirst September 10, 2013; DOI: 10.1158/1078-0432.CCR-13-1253 Clinical Cancer Human Cancer Biology Research Gene Expression Profiling using Nanostring Digital RNA Counting to Identify Potential Target Antigens for Melanoma Immunotherapy Rachel E. Beard, Daniel Abate-Daga, Shannon F. Rosati, Zhili Zheng, John R. Wunderlich, Steven A. Rosenberg, and Richard A. Morgan Abstract Purpose: The success of immunotherapy for the treatment of metastatic cancer is contingent on the identification of appropriate target antigens. Potential targets must be expressed on tumors but show restricted expression on normal tissues. To maximize patient eligibility, ideal target antigens should be expressed on a high percentage of tumors within a histology and, potentially, in multiple different malignancies. Design: A Nanostring probeset was designed containing 97 genes, 72 of which are considered potential candidate genes for immunotherapy. Five established melanoma cell lines, 59 resected metastatic mela- noma tumors, and 31 normal tissue samples were profiled and analyzed using Nanostring technology. Results: Of the 72 potential target genes, 33 were overexpressed in more than 20% of studied melanoma tumor samples. Twenty of those genes were identified as differentially expressed between normal tissues and tumor samples by ANOVA analysis. Analysis of normal tissue gene expression identified seven genes with limited normal tissue expression that warrant further consideration as potential immunotherapy target antigens: CSAG2, MAGEA3, MAGEC2, IL13RA2, PRAME, CSPG4, and SOX10. These genes were highly overexpressed on a large percentage of the studied tumor samples, with expression in a limited number of normal tissue samples at much lower levels. Conclusion: The application of Nanostring RNA counting technology was used to directly quantitate the gene expression levels of multiple potential tumor antigens. -

Seromic Profiling of Ovarian and Pancreatic Cancer
Seromic profiling of ovarian and pancreatic cancer Sacha Gnjatica,1, Erika Rittera, Markus W. Büchlerb, Nathalia A. Gieseb, Benedikt Brorsc, Claudia Freid, Anne Murraya, Niels Halamad, Inka Zörnigd, Yao-Tseng Chene, Christopher Andrewsf, Gerd Rittera, Lloyd J. Olda,1, Kunle Odunsig,2, and Dirk Jägerd,2 aLudwig Institute for Cancer Research Ltd, Memorial-Sloan Kettering Cancer Center, New York, NY 10065; bDepartment of General Surgery, cDepartment of Theoretical Bioinformatics, and dMedizinische Onkologie, Nationales Centrum für Tumorerkrankungen, University Hospital Heidelberg, Heidelberg D-69120, Germany; eDepartment of Pathology, Weill Medical College of Cornell University, New York, NY 10065; and fDepartment of Biostatistics and gDepartment of Gynecologic Oncology, Roswell Park Cancer Institute, Buffalo, NY 14263 Contributed by Lloyd J. Old, December 10, 2009 (sent for review August 20, 2009) Autoantibodies, a hallmark of both autoimmunity and cancer, analyzing a series of lung cancer and healthy control sera on a represent an easily accessible surrogate for measuring adaptive small array (329 proteins) for antigen reactivity using this anti- immune responses to cancer. Sera can now be assayed for re- body profiling method, referred to here as “seromics,” we were activity against thousands of proteins using microarrays, but there able to detect known antigens with sensitivity and specificity is no agreed-upon standard to analyze results. We developed a set comparable to ELISA, as well as new antigens that are now of tailored quality control and normalization procedures based on under further investigation. Contrary to gene microarrays where ELISA validation to allow patient comparisons and determination changes in the pattern of gene expression are detected in clus- of individual cutoffs for specificity and sensitivity. -

Radioallergosorbent Test (RAST)—Reliable Tool Or Poor Substitute?
Radioallergosorbent test (RAST) VII Ö1 1 reliable tool or poor substitute? Edward W. Hein, M.D. An in vitro method, the radioallergosorbent test (RAST) has been developed for the detection of allergen-specific antibodies of the IgE class. Review of the literature shows that in comparison to skin testing, the RAST has a high degree of correlation (60% to 90% depending on the antigen); however, this method is not as sensitive as other tests (50% false-negative). The RAST is affected by blocking antibodies (IgG), resulting in false-negative values and high levels of IgE that bind on the allergen discs, giving false- positive findings. Because of these problems, RAST is somewhat limited for use in the clinical setting. Index terms: Allergy and immunology • Radioallergosor- bent test (RAST) Cleve Clin Q 50:361-366, Fall 1983 Skin testing has been the traditional method for diagnosis of IgE-mediated allergic disorders. This bioassay is highly sensitive, cost-effective, and safe when used by experienced personnel. In 1967, Wide et al,1 in Sweden, reported a new technique capable of detecting the minute quantities of allergen-specific IgE antibodies that circulate in the serum of allergic patients. This laboratory procedure, called RAST (radioallergosorbent test), utilized a solid- phase radioimmunoassay method. During the last decade this in vitro test has been refined and is now a commercially 1 Departments of Pediatric and Adolescent Medi- available laboratory test for clinical laboratories and, in kit cine, and Allergy and Immunology, The Cleveland form, for physicians' offices. Proponents of this new Clinic Foundation. (E.W.H., Head, Section Pediatric method claim that its results are more objective, safer for Allergy and Clinical Immunology.) Submitted for pub- lication May 1983; accepted June 1983. -

Hypersensitivity Reactions (Types I, II, III, IV)
Hypersensitivity Reactions (Types I, II, III, IV) April 15, 2009 Inflammatory response - local, eliminates antigen without extensively damaging the host’s tissue. Hypersensitivity - immune & inflammatory responses that are harmful to the host (von Pirquet, 1906) - Type I Produce effector molecules Capable of ingesting foreign Particles Association with parasite infection Modified from Abbas, Lichtman & Pillai, Table 19-1 Type I hypersensitivity response IgE VH V L Cε1 CL Binds to mast cell Normal serum level = 0.0003 mg/ml Binds Fc region of IgE Link Intracellular signal trans. Initiation of degranulation Larche et al. Nat. Rev. Immunol 6:761-771, 2006 Abbas, Lichtman & Pillai,19-8 Factors in the development of allergic diseases • Geographical distribution • Environmental factors - climate, air pollution, socioeconomic status • Genetic risk factors • “Hygiene hypothesis” – Older siblings, day care – Exposure to certain foods, farm animals – Exposure to antibiotics during infancy • Cytokine milieu Adapted from Bach, JF. N Engl J Med 347:911, 2002. Upham & Holt. Curr Opin Allergy Clin Immunol 5:167, 2005 Also: Papadopoulos and Kalobatsou. Curr Op Allergy Clin Immunol 7:91-95, 2007 IgE-mediated diseases in humans • Systemic (anaphylactic shock) •Asthma – Classification by immunopathological phenotype can be used to determine management strategies • Hay fever (allergic rhinitis) • Allergic conjunctivitis • Skin reactions • Food allergies Diseases in Humans (I) • Systemic anaphylaxis - potentially fatal - due to food ingestion (eggs, shellfish, -

Genome-Wide Analysis of DNA Methylation, Copy Number Variation, and Gene Expression in Monozygotic Twins Discordant for Primary Biliary Cirrhosis
UC Davis UC Davis Previously Published Works Title Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis. Permalink https://escholarship.org/uc/item/34d4m5nk Journal Frontiers in immunology, 5(MAR) ISSN 1664-3224 Authors Selmi, Carlo Cavaciocchi, Francesca Lleo, Ana et al. Publication Date 2014 DOI 10.3389/fimmu.2014.00128 Peer reviewed eScholarship.org Powered by the California Digital Library University of California ORIGINAL RESEARCH ARTICLE published: 28 March 2014 doi: 10.3389/fimmu.2014.00128 Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis Carlo Selmi 1,2*, Francesca Cavaciocchi 1,3, Ana Lleo4, Cristina Cheroni 5, Raffaele De Francesco5, Simone A. Lombardi 1, Maria De Santis 1,3, Francesca Meda1, Maria Gabriella Raimondo1, Chiara Crotti 1, Marco Folci 1, Luca Zammataro1, Marlyn J. Mayo6, Nancy Bach7, Shinji Shimoda8, Stuart C. Gordon9, Monica Miozzo10,11, Pietro Invernizzi 4, Mauro Podda1, Rossana Scavelli 5, Michelle R. Martin12, Michael F. Seldin13,14, Janine M. LaSalle 12 and M. Eric Gershwin2 1 Division of Rheumatology and Clinical Immunology, Humanitas Clinical and Research Center, Milan, Italy 2 Division of Rheumatology, Allergy, and Clinical Immunology, University of California at Davis, Davis, CA, USA 3 BIOMETRA Department, University of Milan, Milan, Italy 4 Liver Unit and Center for Autoimmune Liver Diseases, Humanitas Clinical and Research Center, Milan, Italy 5 National Institute of Molecular Genetics (INGM), Milan, Italy 6 University of Texas Southwestern, Dallas, TX, USA 7 Mt. Sinai University, NewYork, NY, USA 8 Clinical Research Center, National Nagasaki Medical Center, Nagasaki, Japan 9 Henry Ford Hospital, Detroit, MI, USA 10 Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy 11 Division of Pathology, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milan, Italy 12 Genome Center and M.I.N.D.