Chew Et Al-2021-Nature Communi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Alu Element-Based Model of Human Genome Instability George Wyndham Cook, Jr
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2013 An Alu element-based model of human genome instability George Wyndham Cook, Jr. Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Recommended Citation Cook, Jr., George Wyndham, "An Alu element-based model of human genome instability" (2013). LSU Doctoral Dissertations. 2090. https://digitalcommons.lsu.edu/gradschool_dissertations/2090 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. AN ALU ELEMENT-BASED MODEL OF HUMAN GENOME INSTABILITY A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Biological Sciences by George Wyndham Cook, Jr. B.S., University of Arkansas, 1975 May 2013 TABLE OF CONTENTS LIST OF TABLES ...................................................................................................... iii LIST OF FIGURES .................................................................................................... iv LIST OF ABBREVIATIONS ...................................................................................... -

Cancer Immunity (1 December 2006) Vol
Cancer Immun 1424-9634Academy of Cancer Immunology Cancer Immunity (1 December 2006) Vol. 6, p. 12 Submitted: 26 September 2006. Accepted: 10 October 2006. Copyright © 2006 by Andrew J. G. Simpson 061012 Article Physical interaction of two cancer-testis antigens, MAGE-C1 (CT7) and NY-ESO-1 (CT6) Hearn J. Cho1*,**, Otavia L. Caballero2*, Sacha Gnjatic2, Valéria C. C. Andrade3, Gisele W. Colleoni3, Andre L. Vettore4, Hasina H. Outtz1, Sheila Fortunato2, Nasser Altorki1, Cathy A. Ferrera1, Ramon Chua2, Achim A. Jungbluth2, Yao-Tseng Chen1, Lloyd J. Old2 and Andrew J. G. Simpson2 1Weill Medical College of Cornell University, 1300 York Avenue, New York, NY 10021, USA 2Ludwig Institute for Cancer Research, New York Branch at Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021, USA 3Escola Paulista de Medicina, Universidade Federal de Sao Paulo, Sao Paulo, SP, Brazil 4Ludwig Institute for Cancer Research, Sao Paulo Branch, Sao Paulo, SP, Brazil *These authors contributed equally to this work **Present address: NYU Cancer Institute, New York University School of Medicine, 550 First Avenue, New York, NY 10016, USA Contributed by: LJ Old Cancer/testis (CT) antigens are the protein products of germ line- encoded on the X chromosome (CT-X antigens) and those that associated genes that are activated in a wide variety of tumors and can are not (non-X CT antigens) (1). elicit autologous cellular and humoral immune responses. CT It is estimated that 10% of the genes on the X-chromosome antigens can be divided between those that are encoded on the X belong to CT-X families (5). -

Histone Variants: Deviants?
Downloaded from genesdev.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Histone variants: deviants? Rohinton T. Kamakaka2,3 and Sue Biggins1 1Division of Basic Sciences, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA; 2UCT/National Institutes of Health, Bethesda, Maryland 20892, USA Histones are a major component of chromatin, the pro- sealing the two turns of DNA. The nucleosome filament tein–DNA complex fundamental to genome packaging, is then folded into a 30-nm fiber mediated in part by function, and regulation. A fraction of histones are non- nucleosome–nucleosome interactions, and this fiber is allelic variants that have specific expression, localiza- probably the template for most nuclear processes. Addi- tion, and species-distribution patterns. Here we discuss tional levels of compaction enable these fibers to be recent progress in understanding how histone variants packaged into the small volume of the nucleus. lead to changes in chromatin structure and dynamics to The packaging of DNA into nucleosomes and chroma- carry out specific functions. In addition, we review his- tin positively or negatively affects all nuclear processes tone variant assembly into chromatin, the structure of in the cell. While nucleosomes have long been viewed as the variant chromatin, and post-translational modifica- stable entities, there is a large body of evidence indicat- tions that occur on the variants. ing that they are highly dynamic (for review, see Ka- makaka 2003), capable of being altered in their compo- Supplemental material is available at http://www.genesdev.org. sition, structure, and location along the DNA. Enzyme Approximately two meters of human diploid DNA are complexes that either post-translationally modify the packaged into the cell’s nucleus with a volume of ∼1000 histones or alter the position and structure of the nucleo- µm3. -

Histone H2A Bbd (H2AFB2) (NM 001017991) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP323551 Histone H2A Bbd (H2AFB2) (NM_001017991) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human H2A histone family, member B2 (H2AFB2) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 12.5 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_001017991 Locus ID: 474381 UniProt ID: P0C5Z0 RefSeq Size: 594 Cytogenetics: Xq28 RefSeq ORF: 345 Synonyms: H2A.Bbd; H2AB3; H2AFB2 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Histone H2A Bbd (H2AFB2) (NM_001017991) Human Recombinant Protein – TP323551 Summary: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). -
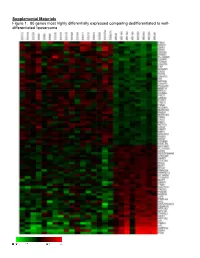
Supplemental Materials Figure 1. 80 Genes Most Highly Differentially Expressed Comparing Dedifferentiated to Well- Differentiated Liposarcoma
Supplemental Materials Figure 1. 80 genes most highly differentially expressed comparing dedifferentiated to well- differentiated liposarcoma Figure 2. Validation of microa CDC2, RACGAP1, FGFR-2, MAD2 and CITED1 200 CDK4 by Liposarcoma Subtype 16 0 12 0 80 Expression40 Level 0 Nl Fat rray data by quantitative RT-P Well Diff 16 0 p16 by Liposarcoma Subtype Dediff 12 0 80 Myxoid Expression40 Level Round Cell 0 12 0 MDM2 by Liposarcoma Subtype 10 0 Pleomorphic Nl Fat 80 60 40 Expression Level 20 RACGAP1 by LiposarcomaWell Diff Subtype 0 70 CR for genes CDK4, MDM2, p16, 60 Dediff 50 Nl Fat 40 30 Myxoid Expression20 Level Well Diff 10 50 0 Round Cell CDC2 by Liposarcoma Subtype Dediff 40 Nl Fat Pleomorphic 30 Myxoid 20 Expression Level Well Diff 10 MAD2L1 by Liposarcoma Subtype Round Cell 60 0 50 Dediff Pleomorphic 40 Nl Fat 30 Myxoid 20 Expression Level Well Diff 10 FGFR-2 by Liposarcoma Subtype 0 Round Cell 200 16 0 Dediff Nl Fat Pleomorphic 12 0 Myxoid 80 Expression Level Well Diff 40 Round Cell 0 Dediff Pleomorphic Nl Fat Myxoid Well Diff CITED1 by Liposarcoma Subtype Round Cell 10 0 80 Dediff Pleomorphic 60 40 Myxoid Expression Level 20 0 Round Cell Nl Fat Pleomorphic Well Diff Dediff Myxoid Round Cell Pleomorphic Table 1. 142 Gene classifier for liposarcoma subtype The genes used for each pair wise subtype comparison are grouped together. The flag column indicates which genes are unique to each subtype comparison. The values show the mean expression levels (actually the mean of the log expression levels was computed and than transformed back to absolute expression level). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Expression of Cancer-Testis Antigens MAGEA1, MAGEA3, ACRBP, PRAME, SSX2, and CTAG2 in Myxoid and Round Cell Liposarcoma
Modern Pathology (2014) 27, 1238–1245 1238 & 2014 USCAP, Inc All rights reserved 0893-3952/14 $32.00 Expression of cancer-testis antigens MAGEA1, MAGEA3, ACRBP, PRAME, SSX2, and CTAG2 in myxoid and round cell liposarcoma Jessica A Hemminger1, Amanda Ewart Toland2, Thomas J Scharschmidt3, Joel L Mayerson3, Denis C Guttridge2 and O Hans Iwenofu1 1Department of Pathology and Laboratory Medicine, Wexner Medical Center at The Ohio State University, Columbus, OH, USA; 2Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University Wexner Medical Center, Columbus, OH, USA and 3Department of Orthopedics, The Ohio State University Wexner Medical Center, Columbus, OH, USA Myxoid and round-cell liposarcoma is a frequently encountered liposarcoma subtype. The mainstay of treatment remains surgical excision with or without chemoradiation. However, treatment options are limited in the setting of metastatic disease. Cancer-testis antigens are immunogenic antigens with the expression largely restricted to testicular germ cells and various malignancies, making them attractive targets for cancer immunotherapy. Gene expression studies have reported the expression of various cancer-testis antigens in liposarcoma, with mRNA expression of CTAG1B, CTAG2, MAGEA9, and PRAME described specifically in myxoid and round-cell liposarcoma. Herein, we further explore the expression of the cancer-testis antigens MAGEA1, ACRBP, PRAME, and SSX2 in myxoid and round-cell liposarcoma by immunohistochemistry in addition to determining mRNA levels of CTAG2 (LAGE-1), PRAME, and MAGEA3 by quantitative real-time PCR. Samples in formalin-fixed paraffin-embedded blocks (n ¼ 37) and frozen tissue (n ¼ 8) were obtained for immunohistochemistry and quantitative real-time PCR, respectively. Full sections were stained with antibodies to MAGEA1, ACRBP, PRAME, and SSX2 and staining was assessed for intensity (1–2 þ ) and percent tumor positivity. -

Network Assessment of Demethylation Treatment in Melanoma: Differential Transcriptome-Methylome and Antigen Profile Signatures
RESEARCH ARTICLE Network assessment of demethylation treatment in melanoma: Differential transcriptome-methylome and antigen profile signatures Zhijie Jiang1☯, Caterina Cinti2☯, Monia Taranta2, Elisabetta Mattioli3,4, Elisa Schena3,5, Sakshi Singh2, Rimpi Khurana1, Giovanna Lattanzi3,4, Nicholas F. Tsinoremas1,6, 1 Enrico CapobiancoID * a1111111111 1 Center for Computational Science, University of Miami, Miami, FL, United States of America, 2 Institute of Clinical Physiology, CNR, Siena, Italy, 3 CNR Institute of Molecular Genetics, Bologna, Italy, 4 IRCCS Rizzoli a1111111111 Orthopedic Institute, Bologna, Italy, 5 Endocrinology Unit, Department of Medical & Surgical Sciences, Alma a1111111111 Mater Studiorum University of Bologna, S Orsola-Malpighi Hospital, Bologna, Italy, 6 Department of a1111111111 Medicine, University of Miami, Miami, FL, United States of America a1111111111 ☯ These authors contributed equally to this work. * [email protected] OPEN ACCESS Abstract Citation: Jiang Z, Cinti C, Taranta M, Mattioli E, Schena E, Singh S, et al. (2018) Network assessment of demethylation treatment in Background melanoma: Differential transcriptome-methylome and antigen profile signatures. PLoS ONE 13(11): In melanoma, like in other cancers, both genetic alterations and epigenetic underlie the met- e0206686. https://doi.org/10.1371/journal. astatic process. These effects are usually measured by changes in both methylome and pone.0206686 transcriptome profiles, whose cross-correlation remains uncertain. We aimed to assess at Editor: Roger Chammas, Universidade de Sao systems scale the significance of epigenetic treatment in melanoma cells with different met- Paulo, BRAZIL astatic potential. Received: June 20, 2018 Accepted: October 17, 2018 Methods and findings Published: November 28, 2018 Treatment by DAC demethylation with 5-Aza-2'-deoxycytidine of two melanoma cell lines Copyright: © 2018 Jiang et al. -

H2A.B Facilitates Transcription Elongation at Methylated Cpg Loci
Downloaded from genome.cshlp.org on October 27, 2017 - Published by Cold Spring Harbor Laboratory Press Research H2A.B facilitates transcription elongation at methylated CpG loci Yibin Chen,1 Qiang Chen,1 Richard C. McEachin,2 James D. Cavalcoli,2 and Xiaochun Yu1,3 1Division of Molecular Medicine and Genetics, Department of Internal Medicine, 2Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA H2A.B is a unique histone H2A variant that only exists in mammals. Here we found that H2A.B is ubiquitously expressed in major organs. Genome-wide analysis of H2A.B in mouse ES cells shows that H2A.B is associated with methylated DNA in gene body regions. Moreover, H2A.B-enriched gene loci are actively transcribed. One typical example is that H2A.B is enriched in a set of differentially methylated regions at imprinted loci and facilitates transcription elongation. These results suggest that H2A.B positively regulates transcription elongation by overcoming DNA methylation in the tran- scribed region. It provides a novel mechanism by which transcription is regulated at DNA hypermethylated regions. [Supplemental material is available for this article.] Histones are nuclear proteins in eukaryotes that package genomic 2004; Eirin-Lopez et al. 2008; Ishibashi et al. 2010; Soboleva et al. DNA into structural units called nucleosomes (Andrews and Luger 2012; Tolstorukov et al. 2012). 2011). A nucleosome consists of a histone octamer with two copies Although H2A.B has been well characterized in vitro, the each of four core histones that are wrapped with ;146 bp of function and localization of H2A.B in the genome are still unclear. -
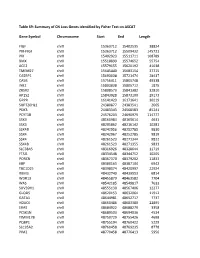
Table S9: Summary of CN Loss Genes Identified by Fisher Test on ASCAT
Table S9: Summary of CN Loss Genes identified by Fisher Test on ASCAT Gene Symbol Chromosome Start End Length FIGF chrX 15363712 15402535 38824 PIR-FIGF chrX 15363712 15509432 145721 PIR chrX 15402923 15511711 108789 BMX chrX 15518899 15574652 55754 ACE2 chrX 15579155 15620192 41038 TMEM27 chrX 15645440 15683154 37715 CA5BP1 chrX 15693038 15721474 28437 CA5B chrX 15756411 15805748 49338 INE2 chrX 15803838 15805712 1875 ZRSR2 chrX 15808573 15841382 32810 AP1S2 chrX 15843928 15873100 29173 GRPR chrX 16141423 16171641 30219 SUPT20HL1 chrX 24380877 24383541 2665 PDK3 chrX 24483343 24568583 85241 PCYT1B chrX 24576203 24690979 114777 SSX9 chrX 48160984 48165614 4631 SSX3 chrX 48205862 48216142 10281 SSX4B chrX 48242956 48252785 9830 SSX4 chrX 48242967 48252785 9819 SSX4 chrX 48261523 48271344 9822 SSX4B chrX 48261523 48271355 9833 SLC38A5 chrX 48316926 48328644 11719 FTSJ1 chrX 48334548 48344752 10205 PORCN chrX 48367370 48379202 11833 EBP chrX 48380163 48387104 6942 TBC1D25 chrX 48398074 48420997 22924 RBM3 chrX 48432740 48439553 6814 WDR13 chrX 48455879 48463582 7704 WAS chrX 48542185 48549817 7633 SUV39H1 chrX 48555130 48567406 12277 GLOD5 chrX 48620153 48632064 11912 GATA1 chrX 48644981 48652717 7737 HDAC6 chrX 48660486 48683380 22895 ERAS chrX 48684922 48688279 3358 PCSK1N chrX 48689503 48694036 4534 TIMM17B chrX 48750729 48755426 4698 PQBP1 chrX 48755194 48760422 5229 SLC35A2 chrX 48760458 48769235 8778 PIM2 chrX 48770458 48776413 5956 OTUD5 chrX 48779302 48815648 36347 KCND1 chrX 48818638 48828251 9614 GRIPAP1 chrX 48830133 48858675 28543 -

The Role of Histone H2av Variant Replacement and Histone H4 Acetylation in the Establishment of Drosophila Heterochromatin
The role of histone H2Av variant replacement and histone H4 acetylation in the establishment of Drosophila heterochromatin Jyothishmathi Swaminathan, Ellen M. Baxter, and Victor G. Corces1 Department of Biology, Johns Hopkins University, Baltimore, Maryland 21218, USA Activation and repression of transcription in eukaryotes involve changes in the chromatin fiber that can be accomplished by covalent modification of the histone tails or the replacement of the canonical histones with other variants. Here we show that the histone H2A variant of Drosophila melanogaster, H2Av, localizes to the centromeric heterochromatin, and it is recruited to an ectopic heterochromatin site formed by a transgene array. His2Av behaves genetically as a PcG gene and mutations in His2Av suppress position effect variegation (PEV), suggesting that this histone variant is required for euchromatic silencing and heterochromatin formation. His2Av mutants show reduced acetylation of histone H4 at Lys 12, decreased methylation of histone H3 at Lys 9, and a reduction in HP1 recruitment to the centromeric region. H2Av accumulation or histone H4 Lys 12 acetylation is not affected by mutations in Su(var)3-9 or Su(var)2-5. The results suggest an ordered cascade of events leading to the establishment of heterochromatin and requiring the recruitment of the histone H2Av variant followed by H4 Lys 12 acetylation as necessary steps before H3 Lys 9 methylation and HP1 recruitment can take place. [Keywords: Chromatin; silencing; transcription; histone; nucleus] Received September 8, 2004; revised version accepted November 4, 2004. The basic unit of chromatin is the nucleosome, which is guchi et al. 2004). The role of histone variants, and spe- made up of 146 bp of DNA wrapped around a histone cially those of H3 and H2A, in various nuclear processes octamer composed of two molecules each of the histones has been long appreciated (Wolffe and Pruss 1996; Ah- H2A, H2B, H3, and H4. -

Histone H2A Bbd (H2AFB2) (NM 001017991) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RG223551 Histone H2A Bbd (H2AFB2) (NM_001017991) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Histone H2A Bbd (H2AFB2) (NM_001017991) Human Tagged ORF Clone Tag: TurboGFP Symbol: H2AB2 Synonyms: H2A.Bbd; H2AB3; H2AFB2 Vector: pCMV6-AC-GFP (PS100010) E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RG223551 representing NM_001017991 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGCCGAGGAGGAGGAGACGCCGAGGGTCCTCCGGTGCTGGCGGCCGGGGGCGGACCTGCTCTCGCACCG TCCGAGCGGAGCTTTCGTTTTCAGTGAGCCAGGTGGAGCGCAGTCTACGGGAGGGCCACTACGCTCAGCG CCTGAGTCGCACGGCGCCGGTCTACCTCGCTGCGGTTATTGAGTACCTGACGGCCAAGGTCCTGGAGCTG GCGGGCAACGAGGCCCAGAACAGCGGAGAGCGGAACATCACTCCCCTGCTGCTGGACATGGTGGTTCACA ACGACAGGCTACTGAGCACCCTTTTCAACACGACCACCATCTCTCAAGTGGCCCCTGGCGAGGAC ACGCGTACGCGGCCGCTCGAG - GFP Tag - GTTTAA Protein Sequence: >RG223551 representing NM_001017991 Red=Cloning site Green=Tags(s) MPRRRRRRGSSGAGGRGRTCSRTVRAELSFSVSQVERSLREGHYAQRLSRTAPVYLAAVIEYLTAKVLEL AGNEAQNSGERNITPLLLDMVVHNDRLLSTLFNTTTISQVAPGED TRTRPLE - GFP Tag - V Restriction Sites: SgfI-MluI This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200,