Elemental and Isotopic Measurements on Palladium After Heavy Water Electrolysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
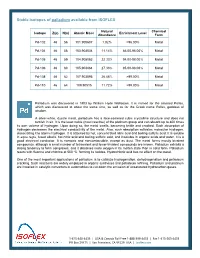
Stable Isotopes of Palladium Available from ISOFLEX
Stable isotopes of palladium available from ISOFLEX Natural Chemical Isotope Z(p) N(n) Atomic Mass Enrichment Level Abundance Form Pd-102 46 56 101.905607 1.02% >96.00% Metal Pd-104 46 58 103.904034 11.14% 86.00-98.00% Metal Pd-105 46 59 104.905083 22.33% 94.00-98.00% Metal Pd-106 46 60 105.903484 27.33% 95.00-98.00% Metal Pd-108 46 62 107.903895 26.46% >99.00% Metal Pd-110 46 64 109.90515 11.72% >99.00% Metal Palladium was discovered in 1803 by William Hyde Wollaston. It is named for the asteroid Pallas, which was discovered at about the same time, as well as for the Greek name Pallas, goddess of wisdom. A silver-white, ductile metal, palladium has a face-centered cubic crystalline structure and does not tarnish in air. It is the least noble (most reactive) of the platinum group and can absorb up to 800 times its own volume of hydrogen. Upon doing so, the metal swells, becoming brittle and cracked. Such absorption of hydrogen decreases the electrical conductivity of the metal. Also, such absorption activates molecular hydrogen, dissociating it to atomic hydrogen. It is attacked by hot, concentrated nitric acid and boiling sulfuric acid. It is soluble in aqua regia, fused alkalis, hot nitric acid and boiling sulfuric acid, and insoluble in organic acids and water. It is a good electrical conductor. It is nontoxic and noncombustible, except as dust. The metal forms mostly bivalent compounds, although a small number of tetravalent and fewer trivalent compounds are known. -

(12) United States Patent (10) Patent No.: US 8,625,731 B2 Holden Et Al
USOO8625731B2 (12) United States Patent (10) Patent No.: US 8,625,731 B2 Holden et al. (45) Date of Patent: Jan. 7, 2014 (54) COMPACT NEUTRONGENERATOR FOR (51) Int. Cl. MEDICAL AND COMMERCIAL SOTOPE G2G 4/02 (2006.01) PRODUCTION, FISSION PRODUCT G2G 4/00 (2006.01) PURIFICATION AND CONTROLLED (52) U.S. Cl. GAMMA REACTIONS FOR DIRECT USPC ............................ 376/108: 376/112:376/156 ELECTRIC POWER GENERATION (58) Field of Classification Search USPC .......................... 376/157, 108,156, 171, 172 (76) Inventors: Charles S. Holden, San Francisco, CA See application file for complete search history. (US); Robert E. Schenter, Portland, OR (US) (56) References Cited *) Notice: Subject to anyy disclaimer, the term of this U.S. PATENT DOCUMENTS patent is extended or adjusted under 35 3,748,226 A * 7/1973 Ribe et al. ..................... 376,124 U.S.C. 154(b) by 961 days. 3,778,627 A * 12/1973 Carpenter ...... ... 376, 192 4,749,540 A * 6/1988 Bogartet al. .. ... 376/133 (21) Appl. No.: 12/296,844 4,997,619 A * 3/1991 Pettus ............ ... 376,288 2004/02284.33 A1* 1 1/2004 Magill et al. ... 376/347 (22) PCT Filed: Apr. 13, 2007 2005/022O248 A1* 10, 2005 Ritter ...... ... 376/190 2008/O144762 A1* 6/2008 Holden et al. ..... ... 376/416 (86). PCT No.: PCT/US2OOTAO66668 * cited by examiner S371 (c)(1), (2), (4) Date: Oct. 10, 2008 Primary Examiner — Jack W Keith Assistant Examiner — Sean P Burke (87) PCT Pub. No.: WO2008/060663 (74) Attorney, Agent, or Firm — Craig M. Stainbrook; PCT Pub. Date: May 22, 2008 Stainbrook & Stainbrook, LLP (65) Prior Publication Data (57) ABSTRACT A neutron generator and isotope production apparatus and US 2011 FO268237 A1 Nov. -

The Discoverers of the Ruthenium Isotopes
•Platinum Metals Rev., 2011, 55, (4), 251–262• The Discoverers of the Ruthenium Isotopes Updated information on the discoveries of the six platinum group metals to 2010 http://dx.doi.org/10.1595/147106711X592448 http://www.platinummetalsreview.com/ By John W. Arblaster This review looks at the discovery and the discoverers Wombourne, West Midlands, UK of the thirty-eight known ruthenium isotopes with mass numbers from 87 to 124 found between 1931 and 2010. Email: [email protected] This is the sixth and fi nal review on the circumstances surrounding the discoveries of the isotopes of the six platinum group elements. The fi rst review on platinum isotopes was published in this Journal in October 2000 (1), the second on iridium isotopes in October 2003 (2), the third on osmium isotopes in October 2004 (3), the fourth on palladium isotopes in April 2006 (4) and the fi fth on rhodium isotopes in April 2011 (5). An update on the new isotopes of palladium, osmium, iridium and platinum discovered since the previous reviews in this series is also included. Naturally Occurring Ruthenium Of the thirty-eight known isotopes of ruthenium, seven occur naturally with the authorised isotopic abun- dances (6) shown in Table I. The isotopes were fi rst detected in 1931 by Aston (7, 8) using a mass spectrograph at the Cavendish Laboratory, Cambridge University, UK. Because of diffi cult experimental conditions due to the use of poor quality samples, Aston actually only detected six of the isotopes and obtained very approximate Table I The Naturally Occurring Isotopes of Ruthenium Mass number Isotopic Abundance, % 96Ru 5.54 98Ru 1.87 99Ru 12.76 100Ru 12.60 101Ru 17.06 102Ru 31.55 104Ru 18.62 251 © 2011 Johnson Matthey http://dx.doi.org/10.1595/147106711X592448 •Platinum Metals Rev., 2011, 55, (4)• percentage abundances. -

Zerohack Zer0pwn Youranonnews Yevgeniy Anikin Yes Men
Zerohack Zer0Pwn YourAnonNews Yevgeniy Anikin Yes Men YamaTough Xtreme x-Leader xenu xen0nymous www.oem.com.mx www.nytimes.com/pages/world/asia/index.html www.informador.com.mx www.futuregov.asia www.cronica.com.mx www.asiapacificsecuritymagazine.com Worm Wolfy Withdrawal* WillyFoReal Wikileaks IRC 88.80.16.13/9999 IRC Channel WikiLeaks WiiSpellWhy whitekidney Wells Fargo weed WallRoad w0rmware Vulnerability Vladislav Khorokhorin Visa Inc. Virus Virgin Islands "Viewpointe Archive Services, LLC" Versability Verizon Venezuela Vegas Vatican City USB US Trust US Bankcorp Uruguay Uran0n unusedcrayon United Kingdom UnicormCr3w unfittoprint unelected.org UndisclosedAnon Ukraine UGNazi ua_musti_1905 U.S. Bankcorp TYLER Turkey trosec113 Trojan Horse Trojan Trivette TriCk Tribalzer0 Transnistria transaction Traitor traffic court Tradecraft Trade Secrets "Total System Services, Inc." Topiary Top Secret Tom Stracener TibitXimer Thumb Drive Thomson Reuters TheWikiBoat thepeoplescause the_infecti0n The Unknowns The UnderTaker The Syrian electronic army The Jokerhack Thailand ThaCosmo th3j35t3r testeux1 TEST Telecomix TehWongZ Teddy Bigglesworth TeaMp0isoN TeamHav0k Team Ghost Shell Team Digi7al tdl4 taxes TARP tango down Tampa Tammy Shapiro Taiwan Tabu T0x1c t0wN T.A.R.P. Syrian Electronic Army syndiv Symantec Corporation Switzerland Swingers Club SWIFT Sweden Swan SwaggSec Swagg Security "SunGard Data Systems, Inc." Stuxnet Stringer Streamroller Stole* Sterlok SteelAnne st0rm SQLi Spyware Spying Spydevilz Spy Camera Sposed Spook Spoofing Splendide -

Study of the Properties of Hydrogen and Deuterium in Beta Phase Palladium Hydride and Deuteride
Study of the Properties of Hydrogen and Deuterium in Beta Phase Palladium Hydride and Deuteride SIMON ANTHONY STEEL 18/04/2018 Materials and Physics Research Group School of Computing Science & Engineering This thesis is submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy Dedication This work is dedicated to the memory of Professor Donald Keith Ross. “I may not have gone where I intended to go, but I think I have ended up where I needed to be.” Douglas Adams i Contents Dedication .................................................................................................................................... i Figures ...................................................................................................................................... vii Acknowledgements .................................................................................................................... x Glossary .................................................................................................................................... xii Units, Constants, and Standard Values ................................................................................... xiii Conventions in this Document ................................................................................................ xiv Abstract .................................................................................................................................... xv 1 Introduction ........................................................................................................................ -

The Discoverers of the Palladium Isotopes the THIRTY-FOUR KNOWN PALLADIUM ISOTOPES FOUND BETWEEN 1935 and 1997
DOI: 10.1595/147106706X110817 The Discoverers of the Palladium Isotopes THE THIRTY-FOUR KNOWN PALLADIUM ISOTOPES FOUND BETWEEN 1935 AND 1997 By J. W. Arblaster Coleshill Laboratories, Gorsey Lane, Coleshill, West Midlands B46 1JU, U.K.; E-mail: [email protected] This is the fourth in a series of reviews of circumstances surrounding the discoveries of the isotopes of the six platinum group elements. The first review, on platinum isotopes, was published in this Journal in October 2000, the second, on iridium isotopes, was published here in October 2003 and the third, on osmium isotopes, was published in October 2004 (1). The current review looks at the discovery and the discoverers of the thirty-four isotopes of palladium. Of the thirty-four known isotopes of palladium, ment activities found for palladium, such as a half- six occur naturally with the following authorised life of six hours discovered by Fermi et al. in 1934 isotopic abundances (2): (9) and half-lifes of 3 minutes and 60 hours discov- ered by Kurchatov et al. (10) in 1935 do not appear to have been confirmed. The Naturally Occurring Isotopes of Palladium In 1940, Nishima et al. (11) obtained an unspec- Mass number Isotopic abundance, % ified activity with a half-life of 26 minutes which is also likely to have been 111Pd. The actual half-life of 102Pd 1.02 111 104Pd 11.14 Pd is now known to be 23 minutes, so the differ- 105Pd 22.33 ent values obtained above are probably indicative 106 Pd 27.33 of calibration problems. 108 Pd 26.46 These unspecified activities raise problems con- 110Pd 11.72 cerning the precedence for treating each discovery in this paper. -

110 Journal of New Energy Vol. 2, No 2
110 Journal of New Energy Vol. 2, no 2 OPERATING THE LENT-1 TRANSMUTATION REACTOR: A PRELIMINARY REPORT By Hal Fox and Shang-Xian Jin ABSTRACT The Low-Energy Nuclear Transmutation (LENT-1) reactor can transmute thorium into smaller mass elements. This transmutation process differs markedly from the natural decay of thorium-232 into lead-208. Using a small amount of thorium nitrate dissolved in distilled water as the electrolyte, the LENT-1 reactor will transmute essentially all of the thorium into small mass elements in thirty minutes processing time. Considerable development work is required to understand the role of reactor parameters in producing various transmuted smaller mass elements. A. INTRODUCTION Most of the current models of nuclear reactions require that high energy be used to cause nuclear reactions, except for the decay of naturally radioactive substances such as thorium and uranium. Nearly all of the nuclear experimental data has been obtained by experiments based on nuclear reactors or using high energy particle accelerators. The study of nuclear reactions in or on the surface of a metal lattice is relatively new. Two international conferences on Low-Energy Nuclear Reactions have been held and the proceedings published in the Journal of New Energy [1,2]. Several important papers have reported on experiments in which low-energy nuclear reactions are observed. This paper reports on the results that have been achieved by various workers using the Low-Energy Nuclear Transmutation (LENT-1) reactor. The LENT-1 reactor consists of a cylindrical electrode and a disk-shaped electrode positioned on the interior of the cylindrical electrode. -

Stable Isotope Separation in Calutrons
Printed ill tho llnitzd Statss ot 4mcm 4vailsble frurii Nations' Technical Information Service U S :3ep?rtmelli uf Commerce 5285 Port 4oyal Cosd Spririizflald Viryinis 22161 NTlS ptwiodes P~III~~Copy ,498, Microfiche P,C1 I his rcpoti \*!is prspxed as an account of >,dSi-k sponsored by a3 agsncy of the United Sraies Governr;,ent. Neither thc !J nited Skiics i<n.~cti $IK~: nor any agency th2reof. no? any of thzir employoes, (ciakc?.Aijy ivarranty, express or irripiled, or assunias any legal liability or responsibility for ihe accuracy. compleienass, or usefulness of any inforriraiton. ;p ius, product, or process disclosed, or represents that its usewould not infr privately o-v?d ilghts Rcferencc herein to ai-y specific commercial product, process, or service by trade nziile, Zi'adei*lark, manufacturer, or otherwise, does not necsssarily constitute or imply its enduisemcat, recorn,n faVOiii-,g by ille United States(;ovc;riiiie!ii 01- any Zgency thereof Th opinions of auiihols cxp:essed here:!: d~ ~iui neccssarily statc or refisc?!host of the United Sta?ezGovernmer:?or any ngcncy therccf. ____ ___________... ..... .____ ORNL/TM-10356 OPERATIONS DIVISION STABLE ISOTOPE SEPARATION IN CALUTRONS: FORTY YEARS OF PRODUCTION AND DISTRIBUTION W. A. Bell J. G. Tracy Date Published: November 1987 Prepared by the OAK RIDGE NATIONAL LABORATORY Oak Ridge, Tennessee 37831 operated by MARTIN MARIETTA ENERGY SYSTEMS, INC. for the U.S. DEPARTMENT OF ENERGY under Contract No. DE-AC05-840R21400 iti CONTENTS Page ACKNOWLEDGMENTS ................................................. iv I. INTRODUCTION .............................................. 1 11. PROGRAM EVOLUTION ......................................... 1 111. RESEARCH AND DEVELOPMENT .................................. 5 IV. CHEMISTRY ................................................. 8 V. -

EPRI. Proceedings: Fourth International Conference on Cold Fusion Volume 3: Nuclear Measurements Papers, TR-104188-V3
EPRI. Proceedings: Fourth International Conference on Cold Fusion Volume 3: Nuclear Measurements Papers, TR-104188-V3. 1994. Lahaina, Maui, Hawaii: Electric Power Research Institute. This book is available here: http://my.epri.com/portal/server.pt?Abstract_id=TR-104188-V3 Product ID: TR-104188-V3 Sector Name: Nuclear Date Published: 7/28/1994 Document Type: Technical Report File size: 18.87 MB File Type: Adobe PDF (.pdf) Full list price: No Charge This Product is publicly available The LENR-CANR version of the book (this file) is 10 MB, and it is in “text under image” or “searchable” Acrobat format. Keywords: EPRI TR-104188-V1 Deuterium Project 3170 EPRI Palladium Proceedings Electric Power Cold fusion July 1994 Research Institute Electrolysis Heat Heavy water Proceedings: Fourth International Conference on Cold Fusion Volume 3: Nuclear Measurements Papers Prepared by Electric Power Research institute Palo Alto, California Proceedings: Fourth International Conference on Cold Fusion Volume 3: Nuclear and Measurements Papers TR-1041 88-V3 Proceedings, July 1994 December 6-9, 1993 Lahaina, Maui, Hawaii Conference Co-chairmen T.O. Passell Electric Power Research Institute M.C.H. McKubre SRI International Prepared by ELECTRIC POWER RESEARCH INSTITUTE 3412 Hillview Avenue Palo Alto, California 94304 Sponsored by Electric Power Research Institute Palo Alto, California T.O. Passel! Nuclear Power Group and Office of Naval Research Arlington, Virginia R. Nowak DISCLAIMER OF WARRANTIES AND LIMITATION OF LIABILITIES THIS REPORT WAS PREPARED BY THE -

Co-Production of Light and Heavy $ R $-Process Elements Via Fission
Draft version June 22, 2020 Typeset using LATEX twocolumn style in AASTeX62 Co-production of Light and Heavy r-process Elements via Fission Deposition Nicole Vassh,1 Matthew R. Mumpower,2, 3 Gail C. McLaughlin,4 Trevor M. Sprouse,5 and Rebecca Surman5, 3 1University of Notre Dame, Notre Dame, IN 46556, USA 2Theoretical Division, Los Alamos National Lab, Los Alamos, NM 87545, USA 3Joint Institute for Nuclear Astrophysics - Center for the Evolution of the Elements, USA 4North Carolina State University, Raleigh, NC 27695, USA 5University of Notre Dame, Notre Dame, Indiana 46556, USA ABSTRACT We apply for the first time fission yields determined across the chart of nuclides from the macroscopic- microscopic theory of the Finite Range Liquid Drop Model to simulations of rapid neutron capture (r-process) nucleosynthesis. With the fission rates and yields derived within the same theoretical framework utilized for other relevant nuclear data, our results represent an important step toward self- consistent applications of macroscopic-microscopic models in r-process calculations. The yields from this model are wide for nuclei with extreme neutron excess. We show that these wide distributions of neutron-rich nuclei, and particularly the asymmetric yields for key species that fission at late times in the r process, can contribute significantly to the abundances of the lighter heavy elements, specifically the light precious metals palladium and silver. Since these asymmetric yields correspondingly also deposit into the lanthanide region, we consider the possible evidence for co-production by comparing our nucleosynthesis results directly with the trends in the elemental ratios of metal-poor stars rich in r-process material. -

Book of Abstracts Lxviii Internatio Nal Conference
MINISTRY OF EDUCATION AND SCIENCE OF RUSSIA RUSSIAN ACADEMY OF SCIENCES JOINT INSTITUTE FOR NUCLEAR RESEARCH SKOBELTSYN INSTITUTE OF NUCLEAR PHYSICS SAINT-PETERSBURG STATE UNIVERSITY VORONEZH STATE UNIVERSITY NOVOVORONEZH NUCLEAR POWER PLANT LXVIII INTERNATIONAL CONFERENCE «NUCLEUS 2018» FUNDAMENTAL PROBLEMS OF NUCLEAR PHYSICS, ATOMIC POWER ENGINEERING AND NUCLEAR TECHNOLOGIES DEDICATED TO THE CENTENNIAL OF VORONEZH STATE UNIVERSITY AND TO THE 80th ANNIVERSARY OF THE BIRTH OF K. A. GRIDNEV BOOK OF ABSTRACTS July 2 – 6, 2018 Voronezh Russia Saint-Petersburg 2018 LXVIII INTERNATIONAL CONFERENCE «NUCLEUS 2018». FUNDAMENTAL PROBLEMS OF NUCLEAR PHYSICS, ATOMIC POWER ENGINEERING AND NUCLEAR TECHNOLOGIES (LXVIII MEETING ON NUCLEAR SPECTROSCOPY AND NUCLEAR STRUCTURE). BOOK OF ABSTRACS. Editor A.K. Vlasnikov The scientific program of the conference covers almost all problems in nuclear physics and its applications such as: neutron-rich nuclei, nuclei far from stability valley, giant resonances, many-phonon and many-quasiparticle states in nuclei, high-spin and super-deformed states in nuclei, synthesis of -hesuperavy elements, reactions with radioactive nuclear beams, heavy ions, nucleons and elementary particles, fusion and fission of nuclei, many-body problem in nuclear physics, microscopic description of collective and single-particle states in nuclei, non-linear nuclear dynamics, meson and quark degrees of freedom in nuclei, mesoatoms, hypernuclei and other nuclear exotic systems, double beta- decay and neutrino mass problem, interaction of -

Neutron Deficient Isotopes of Rhodium and Palladium
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title Neutron Deficient Isotopes of Rhodium and Palladium Permalink https://escholarship.org/uc/item/9d08g7qb Author Perlman, I. Publication Date 2010-02-04 Peer reviewed eScholarship.org Powered by the California Digital Library University of California " '/ ~ ... UCRL a ?,I/C- C-, { UNIVERSITY OF CALIFORNIA J FOR REFERENCE . NOT TO BE TAKEN FROM THIS ROOM BERKELEY, CALIFORNIA ~... :~" }-I .'; '-1 ....- :> ~. S "P' ~ I--( .-... -~1 >-'-<... S ~ --~ ~o" '-J ..... Special Review of r .. Declassified Reports Authorized by USDOE JK Bratton ':) , 0 -"'\0 • ifier Date '. UNrJEPSITY OF CALIFORNIA RJ1.DIATION LABOR~TORY Cover Sheet Do not remove Each person who space below. Route to Noted by Date Route to Noted by Date =====":::;"TF'~=:::::;:"""'====::=';:;:. =.'=:;;:':"=:"::::_,?";;;-=='rr'=:::::==:'::;;:.=:;:;,::;;:.==t=':::;::1=====":= -- . ,.,= "..-1--I ',.",.r----+tII -..----.~.___.._-,_. _.__-......-..\--__-.- -----1---------".....-+1.....· ............-' ,------1--'--- .1 ---..-+---.---.....--f---- I .--+i.__....._~._-----.--f-,--------......-l- .........- .......- f ~.L:i=:Tr;PSl TY OF CALIFORNIA Radiation Laboratory Contract No. W-7405-eng-48 NEUTRON DEFICIEI\lT I SOTOPii;S OF RHODI U!\~ AFD PALLADIUM by Manfred Lindner and I. Perlman February 2, 1948 Berkeley, California ..2- neutron Defid.ent Isotopes of B."odinm. and Palladium Hanfred Lindner and 1. Perlman '1 r' Radiation Laboratory and Department of Chemistr;)r Universitjr of Cal:i.fornia, Berkeley, Ca1iforn:i.a ABSTRACT A <i-hour palladium activit;)r assigned to mass 101 and 4.0 daJr activitJr assicued to mass 100 have been identified. Contract l~o. W-7405-eng-48 To be published as a letter to the Editor of Physical ~~ .'f'........... UCR.j M- Februa_ ~3.