The Radiochemistry of Palladium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Palladium-Catalyzed Vinylic Substitution of Aryl Halides on Polymeric Nitrogen Supports
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1986 Palladium-Catalyzed Vinylic Substitution of Aryl Halides on Polymeric Nitrogen Supports. Chia-hsing Sun Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Sun, Chia-hsing, "Palladium-Catalyzed Vinylic Substitution of Aryl Halides on Polymeric Nitrogen Supports." (1986). LSU Historical Dissertations and Theses. 4268. https://digitalcommons.lsu.edu/gradschool_disstheses/4268 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This reproduction was made from a copy of a manuscript sent to us for publication and microfilming. While the most advanced technology has been used to pho tograph and reproduce this manuscript, the quality of the reproduction is heavily dependent upon the quality of the material submitted. Pages in any manuscript may have indistinct print. In all cases the best available copy has been filmed. The following explanation of techniques is provided to help clarify notations which may appear on this reproduction. 1. Manuscripts may not always be complete. When it is not possible to obtain missing pages, a note appears to Indicate this. 2. When copyrighted materials are removed from the manuscript, a note ap pears to indicate this. 3. Oversize materials (maps, drawings, and charts) are photographed by sec tioning the original, beginning at the upper left hand comer and continu ing from left to right in equal sections with small overlaps. -

Colloidal Goldgold
ColloidalColloidal GoldGold Markus Niederberger Email: [email protected] 22.11.2006 OutlineOutline 1) Definition 2) History 3) Synthesis 4) Chemical and Physical Properties 5) Applications 6) References DefinitionDefinition Colloidal gold: Stable suspension of sub-micrometer- sized particles of gold in a liquid ShortShort HistoryHistory ofof GoldGold 4000 B.C.: A culture in Eastern Europe begins to use gold to fashion decorative objects 2500 B.C.: Gold jewelry was found in the Tomb of Djer, king of the First Egyptian Dynasty 1200 B.C.: The Egyptians master the art of beating gold into leaf as well as alloying it with other metals for hardness and color variations 1091 B.C.: Little squares of gold are used in China as a form of money 300 B.C.: Greeks and Jews of ancient Alexandria start to practice Alchemy, the quest of turning base metals into gold 200 B.C.: The Romans gain access to the gold mining region of Spain 50 B.C.: The Romans begin issuing a gold coin called the Aureus 1284 A.D.: Venice introduces the gold Ducat, which soon becomes the most popular coin in the world Source: National Mining Association, Washington HistoryHistory ofof GoldGold ColloidsColloids andand theirtheir ApplicationApplication History: Gold in Medicine Humankind has linked the lustre of gold with the warm, life-giving light of the sun. In cultures, which deified the sun, gold represented its earthly form. The earliest records of the use of gold for medicinal and healing purposes come from Alexandria, Egypt. Over 5000 years ago the Egyptians ingested gold for mental and bodily purification. -
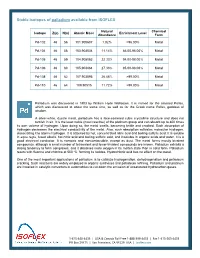
Stable Isotopes of Palladium Available from ISOFLEX
Stable isotopes of palladium available from ISOFLEX Natural Chemical Isotope Z(p) N(n) Atomic Mass Enrichment Level Abundance Form Pd-102 46 56 101.905607 1.02% >96.00% Metal Pd-104 46 58 103.904034 11.14% 86.00-98.00% Metal Pd-105 46 59 104.905083 22.33% 94.00-98.00% Metal Pd-106 46 60 105.903484 27.33% 95.00-98.00% Metal Pd-108 46 62 107.903895 26.46% >99.00% Metal Pd-110 46 64 109.90515 11.72% >99.00% Metal Palladium was discovered in 1803 by William Hyde Wollaston. It is named for the asteroid Pallas, which was discovered at about the same time, as well as for the Greek name Pallas, goddess of wisdom. A silver-white, ductile metal, palladium has a face-centered cubic crystalline structure and does not tarnish in air. It is the least noble (most reactive) of the platinum group and can absorb up to 800 times its own volume of hydrogen. Upon doing so, the metal swells, becoming brittle and cracked. Such absorption of hydrogen decreases the electrical conductivity of the metal. Also, such absorption activates molecular hydrogen, dissociating it to atomic hydrogen. It is attacked by hot, concentrated nitric acid and boiling sulfuric acid. It is soluble in aqua regia, fused alkalis, hot nitric acid and boiling sulfuric acid, and insoluble in organic acids and water. It is a good electrical conductor. It is nontoxic and noncombustible, except as dust. The metal forms mostly bivalent compounds, although a small number of tetravalent and fewer trivalent compounds are known. -

A History of Iridium OVERCOMING the DIFFICULTIES of MELTING and FABRICATION by L
A History of Iridium OVERCOMING THE DIFFICULTIES OF MELTING AND FABRICATION By L. B. Hunt The Johnson Matthey Group The use in unmanned space craft of iridium to encapsulate the radio- isotope thermoelectric generators, where temperatures of up to 20OO0C have to be withstood over several years of operation, with possible impact velocities of 90 metres per second, has focused greater attention on the remarkable properties of this member of the platinum group of metals. But these same properties of very high melting point and great mechanical strength have been the cause of difficulties in its melting and fabrication over a long period of years. How these problems were tackled and eventually overcome is described in this article. One of the less well-known members of the For some fifty years after the discovery of platinum group, iridium possesses quite platinum in South America and the early remarkable chemical and physical properties. It investigations of its properties by a number of is not only the most resistant of all metals to English, French, German and Swedish scien- corrosion, insoluble in all mineral acids in- tists, it was not realised that the native platinum cluding aqua regia and unattacked by other they were examining also contained other molten metals or by silicates at high tem- elements. The first to recognise that a small in- peratures, but has a very high melting point soluble residue survived the dissolution of and is the only metal to maintain good native platinum in aqua regia was the French mechanical properties in air at temperatures chemist Joseph Louis Proust, working for a above 1600OC.Its great stability can be gauged time in Madrid under the patronage of King from its physical properties, outlined in the Carlos IV, but he failed to grasp that other Table. -

Material Safety Data Sheet AQUA REGIA, 2-1-5 (NITRIC 20%/HCL 5%)
2290 Avenue A, Bethlehem, PA 18017 Effective Date: 4/16/2010 NON-EMERGENCY TELEPHONE 24-HOUR CHEMTREC EMERGENCY TELEPHONE 610-866-4225 800-424-9300 Material Safety Data Sheet AQUA REGIA, 2-1-5 (NITRIC 20%/HCL 5%) 1. Product Identification: MIXED ACID CONTAINING NITRIC ACID AND HYDROCHLORIC ACID Synonyms: None CAS No: N/A Molecular Weight: N/A Chemical Formula: N/A 2. Composition/Information on Ingredients Ingredient CAS No Percent Hazardous Nitric Acid 7697-37-2 1 - 25% Yes Hydrochloric Acid 7647-01-0 1 – 10% Yes Water 7732-18-5 65 - 98% No 3. Hazards Identification Emergency Overview POISON! DANGER! CORROSIVE! CORROSIVE. LIQUID AND MIST CAUSE SEVERE BURNS TO ALL BODY TISSUE. MAY BE FATAL IF SWALLOWED OR INHALED. INHALATION MAY CAUSE LUNG AND TOOTH DAMAGE. Potential Health Effects Nitric acid is extremely hazardous; it is a corrosive and a poison. In concentrated solutions it is an oxidizer. Hydrochloric acid is a corrosive. Mixtures of nitric acid and hydrochloric acid may form aqua regia releasing toxic nitrosyl chloride (yellow to reddish-brown) gas. Inhalation: Corrosive! Inhalation of vapors can cause breathing difficulties and lead to pneumonia and pulmonary edema, which may be fatal. Other symptoms may include coughing, choking, and irritation of the nose, throat, and respiratory tract. Ingestion: 1 Corrosive! Swallowing nitric acid and hydrochloric acid can cause immediate pain and burns of the mouth, throat, esophagus and gastrointestinal tract. Skin Contact: Corrosive! Can cause redness, pain, and severe skin burns. Concentrated solutions cause deep ulcers and stain skin a yellow or yellow-brown color. Eye Contact: Corrosive! Vapors are irritating and may cause damage to the eyes. -

(12) United States Patent (10) Patent No.: US 8,625,731 B2 Holden Et Al
USOO8625731B2 (12) United States Patent (10) Patent No.: US 8,625,731 B2 Holden et al. (45) Date of Patent: Jan. 7, 2014 (54) COMPACT NEUTRONGENERATOR FOR (51) Int. Cl. MEDICAL AND COMMERCIAL SOTOPE G2G 4/02 (2006.01) PRODUCTION, FISSION PRODUCT G2G 4/00 (2006.01) PURIFICATION AND CONTROLLED (52) U.S. Cl. GAMMA REACTIONS FOR DIRECT USPC ............................ 376/108: 376/112:376/156 ELECTRIC POWER GENERATION (58) Field of Classification Search USPC .......................... 376/157, 108,156, 171, 172 (76) Inventors: Charles S. Holden, San Francisco, CA See application file for complete search history. (US); Robert E. Schenter, Portland, OR (US) (56) References Cited *) Notice: Subject to anyy disclaimer, the term of this U.S. PATENT DOCUMENTS patent is extended or adjusted under 35 3,748,226 A * 7/1973 Ribe et al. ..................... 376,124 U.S.C. 154(b) by 961 days. 3,778,627 A * 12/1973 Carpenter ...... ... 376, 192 4,749,540 A * 6/1988 Bogartet al. .. ... 376/133 (21) Appl. No.: 12/296,844 4,997,619 A * 3/1991 Pettus ............ ... 376,288 2004/02284.33 A1* 1 1/2004 Magill et al. ... 376/347 (22) PCT Filed: Apr. 13, 2007 2005/022O248 A1* 10, 2005 Ritter ...... ... 376/190 2008/O144762 A1* 6/2008 Holden et al. ..... ... 376/416 (86). PCT No.: PCT/US2OOTAO66668 * cited by examiner S371 (c)(1), (2), (4) Date: Oct. 10, 2008 Primary Examiner — Jack W Keith Assistant Examiner — Sean P Burke (87) PCT Pub. No.: WO2008/060663 (74) Attorney, Agent, or Firm — Craig M. Stainbrook; PCT Pub. Date: May 22, 2008 Stainbrook & Stainbrook, LLP (65) Prior Publication Data (57) ABSTRACT A neutron generator and isotope production apparatus and US 2011 FO268237 A1 Nov. -

Palladium-Catalysed Oxidation of Primary and Secondary Alcohols
Tetrahedron 59 (2003) 5789–5816 Tetrahedron report number 645 Palladium-catalysed oxidation of primary and secondary alcohols Jacques Muzart* Unite´ Mixte de Recherche ‘Re´actions Se´lectives et Applications’, CNRS-Universite´ de Reims Champagne-Ardenne, B.P. 1039, 51687 Reims Cedex 2, France Received 23 May 2003 Contents 1. Introduction 5789 2. Oxygen as a co-oxidant 5790 2.1. Using well-determined PdII-compounds 5790 2.2. Using soluble Pd0 complexes 5795 2.3. Using stabilised palladium particles 5796 2.4. Using supported Pd0 as the starting catalyst 5797 3. Per-compounds as co-oxidants 5798 3.1. tert-Butyl hydroperoxide 5798 3.2. Hydrogen peroxide 5798 3.3. Potassium periodate 5798 4. Halogen-based co-oxidants 5799 4.1. Aromatic halides 5799 4.2. Vinylic bromides 5802 4.3. Carbon tetrachloride 5803 4.4. 1,2-Dichloroethane 5803 4.5. N-Halosuccinimides 5804 5. Metal salts as co-oxidants 5804 6. Allyl-X as hydrogen acceptors 5804 7. Double bonds as hydrogen acceptors 5805 8. Oxidations without co-oxidants or hydrogen acceptors 5808 9. Conclusions 5809 10. Note added in proof 5809 1. Introduction reactions in organic synthesis. Traditionally, stoichiometric and even over-stoichiometric amounts of metal oxides The transformation of the hydroxy group into the corre- (Eq. (1)) or metal salts (Eq. (2)) are used for such oxidations. sponding carbonyl is one of the most frequently used These procedures, which generate copious quantities of heavy metal wastes, are hardly compatible with environ- Keywords: oxidation; dehydrogenation; alcohol; palladium; catalysis. mental regulations. A number of metal-catalysed methods have been described as interesting alternatives; this has Abbreviations: ADP, allyl diethyl phosphate; Adogen, 464w, introduced a third type of reaction (Eq. -

The Discoverers of the Ruthenium Isotopes
•Platinum Metals Rev., 2011, 55, (4), 251–262• The Discoverers of the Ruthenium Isotopes Updated information on the discoveries of the six platinum group metals to 2010 http://dx.doi.org/10.1595/147106711X592448 http://www.platinummetalsreview.com/ By John W. Arblaster This review looks at the discovery and the discoverers Wombourne, West Midlands, UK of the thirty-eight known ruthenium isotopes with mass numbers from 87 to 124 found between 1931 and 2010. Email: [email protected] This is the sixth and fi nal review on the circumstances surrounding the discoveries of the isotopes of the six platinum group elements. The fi rst review on platinum isotopes was published in this Journal in October 2000 (1), the second on iridium isotopes in October 2003 (2), the third on osmium isotopes in October 2004 (3), the fourth on palladium isotopes in April 2006 (4) and the fi fth on rhodium isotopes in April 2011 (5). An update on the new isotopes of palladium, osmium, iridium and platinum discovered since the previous reviews in this series is also included. Naturally Occurring Ruthenium Of the thirty-eight known isotopes of ruthenium, seven occur naturally with the authorised isotopic abun- dances (6) shown in Table I. The isotopes were fi rst detected in 1931 by Aston (7, 8) using a mass spectrograph at the Cavendish Laboratory, Cambridge University, UK. Because of diffi cult experimental conditions due to the use of poor quality samples, Aston actually only detected six of the isotopes and obtained very approximate Table I The Naturally Occurring Isotopes of Ruthenium Mass number Isotopic Abundance, % 96Ru 5.54 98Ru 1.87 99Ru 12.76 100Ru 12.60 101Ru 17.06 102Ru 31.55 104Ru 18.62 251 © 2011 Johnson Matthey http://dx.doi.org/10.1595/147106711X592448 •Platinum Metals Rev., 2011, 55, (4)• percentage abundances. -

Zerohack Zer0pwn Youranonnews Yevgeniy Anikin Yes Men
Zerohack Zer0Pwn YourAnonNews Yevgeniy Anikin Yes Men YamaTough Xtreme x-Leader xenu xen0nymous www.oem.com.mx www.nytimes.com/pages/world/asia/index.html www.informador.com.mx www.futuregov.asia www.cronica.com.mx www.asiapacificsecuritymagazine.com Worm Wolfy Withdrawal* WillyFoReal Wikileaks IRC 88.80.16.13/9999 IRC Channel WikiLeaks WiiSpellWhy whitekidney Wells Fargo weed WallRoad w0rmware Vulnerability Vladislav Khorokhorin Visa Inc. Virus Virgin Islands "Viewpointe Archive Services, LLC" Versability Verizon Venezuela Vegas Vatican City USB US Trust US Bankcorp Uruguay Uran0n unusedcrayon United Kingdom UnicormCr3w unfittoprint unelected.org UndisclosedAnon Ukraine UGNazi ua_musti_1905 U.S. Bankcorp TYLER Turkey trosec113 Trojan Horse Trojan Trivette TriCk Tribalzer0 Transnistria transaction Traitor traffic court Tradecraft Trade Secrets "Total System Services, Inc." Topiary Top Secret Tom Stracener TibitXimer Thumb Drive Thomson Reuters TheWikiBoat thepeoplescause the_infecti0n The Unknowns The UnderTaker The Syrian electronic army The Jokerhack Thailand ThaCosmo th3j35t3r testeux1 TEST Telecomix TehWongZ Teddy Bigglesworth TeaMp0isoN TeamHav0k Team Ghost Shell Team Digi7al tdl4 taxes TARP tango down Tampa Tammy Shapiro Taiwan Tabu T0x1c t0wN T.A.R.P. Syrian Electronic Army syndiv Symantec Corporation Switzerland Swingers Club SWIFT Sweden Swan SwaggSec Swagg Security "SunGard Data Systems, Inc." Stuxnet Stringer Streamroller Stole* Sterlok SteelAnne st0rm SQLi Spyware Spying Spydevilz Spy Camera Sposed Spook Spoofing Splendide -

Study of the Properties of Hydrogen and Deuterium in Beta Phase Palladium Hydride and Deuteride
Study of the Properties of Hydrogen and Deuterium in Beta Phase Palladium Hydride and Deuteride SIMON ANTHONY STEEL 18/04/2018 Materials and Physics Research Group School of Computing Science & Engineering This thesis is submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy Dedication This work is dedicated to the memory of Professor Donald Keith Ross. “I may not have gone where I intended to go, but I think I have ended up where I needed to be.” Douglas Adams i Contents Dedication .................................................................................................................................... i Figures ...................................................................................................................................... vii Acknowledgements .................................................................................................................... x Glossary .................................................................................................................................... xii Units, Constants, and Standard Values ................................................................................... xiii Conventions in this Document ................................................................................................ xiv Abstract .................................................................................................................................... xv 1 Introduction ........................................................................................................................ -

Pd-H from Pd/C and Triethylamine: Implications in Palladium Catalysed Reactions Involving Amines Yoann Coquerel, Paul Brémond, Jean Rodriguez
Pd-H from Pd/C and Triethylamine: Implications in Palladium Catalysed Reactions Involving Amines Yoann Coquerel, Paul Brémond, Jean Rodriguez To cite this version: Yoann Coquerel, Paul Brémond, Jean Rodriguez. Pd-H from Pd/C and Triethylamine: Implications in Palladium Catalysed Reactions Involving Amines. Journal of Organometallic Chemistry, Elsevier, 2007, 692 (22), pp.4805-4808. 10.1016/j.jorganchem.2007.05.053. hal-00676968 HAL Id: hal-00676968 https://hal.archives-ouvertes.fr/hal-00676968 Submitted on 6 Mar 2012 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Pd-H from Pd/C and Triethylamine: Implications in Palladium Catalysed Reactions Involving Amines Yoann Coquerel,* Paul Brémond, Jean Rodriguez* Université Paul Cézanne (Aix-Marseille III), UMR CNRS 6178, Centre universitaire de St Jérôme, Service 531, 13397 Marseille Cedex 20, France. E-mail: [email protected] and jean.rodriguez@univ-cezanne .fr Tel: +33 (0) 491 28 90 88 and +33 (0) 491 28 89 33; Fax: +33 (0) 491 28 91 27 Graphical Abstract Abstract The palladium hydride-iminium complex generated from Pd/C and triethylamine catalyses the isomerisation of allylic alcohols into carbonyl compounds, and Pd/C catalyses the conjugate reduction of activated double bonds using triethylamine as the source of the two newly incorporated hydrogen atoms via the same complex. -

Safe Handling and Disposal of Chemicals Used in the Illicit Manufacture of Drugs
Vienna International Centre, PO Box 500, 1400 Vienna, Austria Tel.: (+43-1) 26060-0, Fax: (+43-1) 26060-5866, www.unodc.org Guidelines for the Safe handling and disposal of chemicals used in the illicit manufacture of drugs United Nations publication USD 26 Printed in Austria ISBN 978-92-1-148266-9 Sales No. E.11.XI.14 ST/NAR/36/Rev.1 V.11-83777—September*1183777* 2011—300 Guidelines for the Safe handling and disposal of chemicals used in the illlicit manufacture of drugs UNITED NATIONS New York, 2011 Symbols of United Nations documents are composed of letters combined with figures. Mention of such symbols indicates a reference to a United Nations document. ST/NAR/36/Rev.1 UNITED NATIONS PUBLICATION Sales No. E.11.XI.14 ISBN 978-92-1-148266-9 eISBN 978-92-1-055160-1 © United Nations, September 2011. All rights reserved. The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. Requests for permission to reproduce this work are welcomed and should be sent to the Secretary of the Publications Board, United Nations Headquarters, New York, N.Y. 10017, U.S.A. or also see the website of the Board: https://unp.un.org/Rights.aspx. Governments and their institutions may reproduce this work without prior authoriza- tion but are requested to mention the source and inform the United Nations of such reproduction.