Identification of a Reaction Intermediate and Mechanism of Action of Intermediary Enzymes in Plumbagin Biosynthetic Pathway Using Molecular Dynamics Simulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

A Phosphopantetheinylating Polyketide Synthase Producing a Linear Polyene to Initiate Enediyne Antitumor Antibiotic Biosynthesis
A phosphopantetheinylating polyketide synthase producing a linear polyene to initiate enediyne antitumor antibiotic biosynthesis Jian Zhang†, Steven G. Van Lanen‡, Jianhua Ju‡, Wen Liu‡, Pieter C. Dorrestein§, Wenli Li‡, Neil L. Kelleher§, and Ben Shen†‡¶ʈ †Department of Chemistry, ‡Division of Pharmaceutical Sciences, and ¶University of Wisconsin National Cooperative Drug Discovery Group, University of Wisconsin, Madison, WI 53705; and §Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, IL 61801 Communicated by Arnold L. Demain, Drew University, Madison, NJ, December 11, 2007 (received for review November 29, 2007) The enediynes, unified by their unique molecular architecture and can have anywhere from all to none of these activities, resulting mode of action, represent some of the most potent anticancer drugs in a product with various levels of oxidation. ever discovered. The biosynthesis of the enediyne core has been Bacterial PKSs are often categorized by domain architecture predicted to be initiated by a polyketide synthase (PKS) that is distinct and the iterative or noniterative nature during polyketide for- from all known PKSs. Characterization of the enediyne PKS involved mation, and this classification has led to three groupings (17). in C-1027 (SgcE) and neocarzinostatin (NcsE) biosynthesis has now Noniterative type I PKS are large proteins with multiple domains revealed that (i) the PKSs contain a central acyl carrier protein domain forming modules, a minimal module consisting of a KS, AT, and and C-terminal phosphopantetheinyl transferase domain; (ii) the PKSs ACP domain, wherein each domain catalyzes a single event are functional in heterologous hosts, and coexpression with an during polyketide biosynthesis (18). -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

(12) Patent Application Publication (10) Pub. No.: US 2011/0086407 A1 Berka Et Al
US 20110086407A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2011/0086407 A1 Berka et al. (43) Pub. Date: Apr. 14, 2011 (54) BACILLUS LCHENFORMS Publication Classification CHROMOSOME (51) Int. Cl. (75) Inventors: Randy Berka, Davis, CA (US); CI2N 9/12 (2006.01) Michael Rey, Davis, CA (US); CI2N 9/90 (2006.01) Preethi Ramaiya, Walnut Creek, C07K I4/32 (2006.01) CA (US); Jens Tonne Andersen, CI2N 9/00 (2006.01) Naerum (DK); Michael Dolberg CI2N 9/56 (2006.01) Rasmussen, Vallensbaek (DK); CI2N 9/16 (2006.01) Peter Bjarke Olsen, Copenhagen O CI2N 9/10 (2006.01) CI2N 9/88 (2006.01) (DK) CI2N 9/78 (2006.01) (73) Assignees: Novozymes A/S, Bagsvaerd (DK); C07K I4/95 (2006.01) Novozymes, Inc., Davis, CA (US) (52) U.S. Cl. ......... 435/194; 435/233; 530/350: 435/183; (21) Appl. No.: 12/972,306 435/222; 435/196; 435/193; 435/232:435/227 (22) Filed: Dec. 17, 2010 Related U.S. Application Data (57) ABSTRACT The present invention relates to an isolated polynucleotide of (62) Division of application No. 12/322.974, filed on Feb. the complete chromosome of Bacillus licheniformis. The 9, 2009, now Pat. No. 7,863,032, which is a division of present invention also relates to isolated genes of the chro application No. 10/983,128, filedon Nov. 5, 2004, now mosome of Bacillus licheniformis which encode biologically Pat. No. 7,494,798. active Substances and to nucleic acid constructs, vectors, and (60) Provisional application No. 60/535.988, filed on Jan. -

Proteome Analysis of Peroxisomes from Dark-Treated Senescent Arabidopsis Leavesfa
Journal of Integrative JIPB Plant Biology Proteome analysis of peroxisomes from dark-treated senescent Arabidopsis leavesFA † † Ronghui Pan1 , Sigrun Reumann1,2,3,4 , Piotr Lisik3, Stefanie Tietz1, Laura J. Olsen2 and Jianping Hu1,5* 1. MSU-Department of Energy Plant Research Laboratory, Michigan State University, East Lansing, MI 48824, USA 2. Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI, USA 3. Center of Organelle Research, University of Stavanger, N-4021 Stavanger, Norway 4. Department of Plant Biochemistry and Infection Biology, Institute of Plant Science and Microbiology, University of Hamburg, D-22609 Hamburg, Germany 5. Plant Biology Department, Michigan State University, East Lansing, MI 48824, USA † These authors contributed equally to this work *Correspondence: Jianping Hu ([email protected]) doi: 10.1111/jipb.12670 High-Impact Article Abstract Peroxisomes compartmentalize a dynamic suite of a total of 111 peroxisomal proteins and verified the peroxisomal biochemical reactions and play a central role in plant localization for six new proteins with potential roles in fatty metabolism, such as the degradation of hydrogen peroxide, acid metabolism and stress response by in vivo targeting metabolism of fatty acids, photorespiration, and the biosyn- analysis. Metabolic pathways compartmentalized in the three thesis of plant hormones. Plant peroxisomes have been major subtypes of peroxisomes were also compared, which traditionally classified into three major subtypes, and in-depth revealed a higher number of proteins involved in the mass spectrometry (MS)-based proteomics has been per- detoxification of reactive oxygen species in peroxisomes formed to explore the proteome of the two major subtypes from senescent leaves. -

Neocarzinostatin Naphthoate Synthase: an Unique Iterative Type I PKS from Neocarzinostatin Producer Streptomyces Carzinostaticus
FEBS 28398 FEBS Letters 566 (2004) 201–206 Neocarzinostatin naphthoate synthase: an unique iterative type I PKS from neocarzinostatin producer Streptomyces carzinostaticus Basundhara Sthapita, Tae-Jin Oha, Rajan Lamichhanea, Kwangkyoung Lioua, Hei Chan Leea, Chun-Gyu Kimb, Jae Kyung Sohnga,* aInstitute of Biomolecule Reconstruction (iBR), Department of Chemistry, Sun Moon University, #100, Kalsan-ri, Tangjeong-myeon, Asansi, Chung-Nam 336-708, Republic of Korea bDepartment of Pharmaceutical Engineering, Inje University, 607 Obang-dong, Kimhae City, Kyungnam 621-749, Republic of Korea Received 13 March 2004; revised 7 April 2004; accepted 7 April 2004 Available online 28 April 2004 Edited by Stuart Ferguson ketide synthase (PKS) catalyzes a decarboxylative condensa- Abstract Enediyne antibiotics are known for their potent antitumor activities. One such enediyne, neocarzinostatin tion of these compounds into the growing polyketide chain. (NCS), consists of a 1:1 complex of non-peptide chromophore Microbial PKSs are classified into three major types. A type I (1a), and peptide apoprotein. The structurally diverse non- PKS system consists of one or more multifunctional proteins peptide chromophore is responsible for its biological activity. that contain a different active site for each enzyme-catalyzed One of its structural components, the naphthoic acid moiety (2,7- reaction in polyketide carbon chain assembly and modification dihydroxy-5-methyl-1-naphthoic acid, 1d) is synthesized by a [1]. The macrolides of erythromycin and avermectin are ex- polyketide synthase (PKS) pathway through condensing six amples of type I PKS products. Type II PKSs, on the other intact acetate units. The 5.45 kb iterative type I PKS, hand, comprise sets of iteratively used individual proteins for neocarzinostatin naphthoate synthase (NNS), responsible for the production of multicyclic aromatic compounds (e.g., ac- naphthoic acid moiety biosynthesis, shares sequence homology tinorhodin and tetrocenomycin) [2]. -

(MNQ) Biosynthesis in Impatiens Balsamina L. Lian Chee Foong1,2, Jian Yi Chai1, Anthony Siong Hock Ho1, Brandon Pei Hui Yeo3, Yang Mooi Lim4 & Sheh May Tam1*
www.nature.com/scientificreports OPEN Comparative transcriptome analysis to identify candidate genes involved in 2‑methoxy‑1,4‑naphthoquinone (MNQ) biosynthesis in Impatiens balsamina L. Lian Chee Foong1,2, Jian Yi Chai1, Anthony Siong Hock Ho1, Brandon Pei Hui Yeo3, Yang Mooi Lim4 & Sheh May Tam1* Impatiens balsamina L. is a tropical ornamental and traditional medicinal herb rich in natural compounds, especially 2‑methoxy‑1,4‑naphthoquinone (MNQ) which is a bioactive compound with tested anticancer activities. Characterization of key genes involved in the shikimate and 1,4‑dihydroxy‑2‑naphthoate (DHNA) pathways responsible for MNQ biosynthesis and their expression profles in I. balsamina will facilitate adoption of genetic/metabolic engineering or synthetic biology approaches to further increase production for pre‑commercialization. In this study, HPLC analysis showed that MNQ was present in signifcantly higher quantities in the capsule pericarps throughout three developmental stages (early‑, mature‑ and postbreaker stages) whilst its immediate precursor, 2‑hydroxy‑1,4‑naphthoquinone (lawsone) was mainly detected in mature leaves. Transcriptomes of I. balsamina derived from leaf, fower, and three capsule developmental stages were generated, totalling 59.643 Gb of raw reads that were assembled into 94,659 unigenes (595,828 transcripts). A total of 73.96% of unigenes were functionally annotated against seven public databases and 50,786 diferentially expressed genes (DEGs) were identifed. Expression profles of 20 selected genes from four major secondary metabolism pathways were studied and validated using qRT‑PCR method. Majority of the DHNA pathway genes were found to be signifcantly upregulated in early stage capsule compared to fower and leaf, suggesting tissue‑specifc synthesis of MNQ. -

Table 4. V. Cholerae Flexgene ORF Collection
Table 4. V. cholerae FLEXGene ORF collection Reference Clone protein PlasmID clone GenBank Locus tag Symbol accession identifier FLEX clone name accession Product name VC0001 NP_062585 VcCD00019918 FLH200476.01F DQ772770 hypothetical protein VC0002 mioC NP_062586 VcCD00019938 FLH200506.01F DQ772771 mioC protein VC0003 thdF NP_062587 VcCD00019958 FLH200531.01F DQ772772 thiophene and furan oxidation protein ThdF VC0004 yidC NP_062588 VcCD00019970 FLH200545.01F DQ772773 inner membrane protein, 60 kDa VC0005 NP_062589 VcCD00061243 FLH236482.01F DQ899316 conserved hypothetical protein VC0006 rnpA NP_062590 VcCD00025697 FLH214799.01F DQ772774 ribonuclease P protein component VC0007 rpmH NP_062591 VcCD00061229 FLH236450.01F DQ899317 ribosomal protein L34 VC0008 NP_062592 VcCD00019917 FLH200475.01F DQ772775 amino acid ABC transporter, ATP-binding protein VC0009 NP_062593 VcCD00019966 FLH200540.01F DQ772776 amino acid ABC transproter, permease protein VC0010 NP_062594 VcCD00019152 FLH199275.01F DQ772777 amino acid ABC transporter, periplasmic amino acid-binding portion VC0011 NP_062595 VcCD00019151 FLH199274.01F DQ772778 hypothetical protein VC0012 dnaA NP_062596 VcCD00017363 FLH174286.01F DQ772779 chromosomal DNA replication initiator DnaA VC0013 dnaN NP_062597 VcCD00017316 FLH174063.01F DQ772780 DNA polymerase III, beta chain VC0014 recF NP_062598 VcCD00019182 FLH199319.01F DQ772781 recF protein VC0015 gyrB NP_062599 VcCD00025458 FLH174642.01F DQ772782 DNA gyrase, subunit B VC0016 NP_229675 VcCD00019198 FLH199346.01F DQ772783 hypothetical protein -

DISSECTING the ROLE of PEROXISOMES in MODULATING ENVIRONMENTAL STRESS RESPONSE and PHOTOSYNTHESIS by Jiying Li
DISSECTING THE ROLE OF PEROXISOMES IN MODULATING ENVIRONMENTAL STRESS RESPONSE AND PHOTOSYNTHESIS By Jiying Li A DISSERTATION Submitted to Michigan State University in partial fulfillment of the requirements for the degree of Genetics - Doctor of Philosophy 2015 ABSTRACT DISSECTING THE ROLE OF PEROXISOMES IN MODULATING ENVIRONMENTAL STRESS RESPONSE AND PHOTOSYNTHESIS By Jiying Li Peroxisomes are essential organelles that house a wide array of metabolic reactions important for plant growth and development. These organelles also interact with other organelles to support cellular functions. However, our knowledge regarding the role of peroxisomal proteins in various biological processes, including plant stress response and photosynthesis, is still incomplete. To address this question at the systems level, I exploited in silico analysis, mutant screens and in-depth physiological and biochemical characterizations. First, I used microarray data to generate a comprehensive view of transcript level changes for Arabidopsis peroxisomal genes during development and under abiotic and biotic stress conditions. Second, mutants of LON2 protease and the photorespiratory enzyme hydroxypyruvate reductase 1 (HPR1) were identified to have enhanced susceptibility to drought, suggesting the involvement of peroxisomal quality control and photorespiration in drought resistance. Third, I conducted a comprehensive peroxisomal mutant screen, in which 147 mutants of 104 Arabidopsis genes encoding peroxisomal proteins were subjected to an automated screening system, -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -
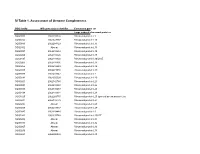
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

(12) United States Patent (10) Patent No.: US 8,168,417 B2 Berka Et Al
USOO81 68417B2 (12) United States Patent (10) Patent No.: US 8,168,417 B2 Berka et al. (45) Date of Patent: May 1, 2012 (54) BACILLUS LICHENIFORMIS CHROMOSOME OTHER PUBLICATIONS Kunst et al., 1997, PIR database Accession No. D69904, “butyrate (75) Inventors: Randy Berka, Davis, CA (US); Michael acetoacetate CoA-transferase (EC 2.8.3.9.) large chain homolog Rey, Davis, CA (US); Preethi Ramaiya, yodR-Bacillus subtilis”.* Walnut Creek, CA (US); Jens Tonne Claus, D. and Berkeley, R.C.W. (1986) in Bergey's Manual of Sys Andersen, Naerum (DK); Michael tematic Bacteriology, vol. 2., eds. Sneath, P.H.A. etal. (Williams and Dolberg Rasmussen, Vallensbaek (DK); Wilkins Co., Baltimore, MD.), pp. 1105-1139. Peter Bjarke Olsen, Copenhagen Ø Eveleigh, D.E. (1981) Scientific American 245, 155-178. (DK) Erickson, R.J. (1976) in Microbiology, ed. Schlesinger, D. (Am. Soc. Microbiol. Washington, DC), pp. 406-419. (73) Assignees: Novozymes A/S, Bagsvaerd (DK); Logan, N. A. and Berkeley, R.C.W. (1981), in The Aerobic Novozymes, Inc., Davis, CA (US) Endospore-Forming Bacteria. Classification and Identification, eds. Berkeley, R.C.W. and Goodfellow, M. (Academic Press, Inc., Lon (*) Notice: Subject to any disclaimer, the term of this don), pp. 106-140. patent is extended or adjusted under 35 O'Donnell, A.G., Norris, J.R., Berkeley, R.C.W., Claus, D., Kanero, U.S.C. 154(b) by 0 days. T., Logan, N. A., and Nozaki, R. (1980) Internat. J. Systematic Bacteriol. 30, 448-459. (21) Appl. No.: 12/972,306 Lapidus et al., (2002), Co-linear scaffold of the Bacillus licheniformis and Bacillus subtilis genomes and its use to compare (22) Filed: Dec.