PFIZER INC. These Results Are Supplied for Informational Purposes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting Fibrosis in the Duchenne Muscular Dystrophy Mice Model: an Uphill Battle
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Targeting fibrosis in the Duchenne Muscular Dystrophy mice model: an uphill battle 2 Marine Theret1#, Marcela Low1#, Lucas Rempel1, Fang Fang Li1, Lin Wei Tung1, Osvaldo 3 Contreras3,4, Chih-Kai Chang1, Andrew Wu1, Hesham Soliman1,2, Fabio M.V. Rossi1 4 1School of Biomedical Engineering and the Biomedical Research Centre, Department of Medical 5 Genetics, 2222 Health Sciences Mall, Vancouver, BC, V6T 1Z3, Canada 6 2Department of Pharmacology and Toxicology, Faculty of Pharmaceutical Sciences, Minia 7 University, Minia, Egypt 8 3Developmental and Stem Cell Biology Division, Victor Chang Cardiac Research Institute, 9 Darlinghurst, NSW, 2010, Australia 10 4Departamento de Biología Celular y Molecular and Center for Aging and Regeneration (CARE- 11 ChileUC), Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, 8331150 12 Santiago, Chile 13 # Denotes Co-first authorship 14 15 Keywords: drug screening, fibro/adipogenic progenitors, fibrosis, repair, skeletal muscle. 16 Correspondence to: 17 Marine Theret 18 School of Biomedical Engineering and the Biomedical Research Centre 19 University of British Columbia 20 2222 Health Sciences Mall, Vancouver, British Columbia 21 Tel: +1(604) 822 0441 fax: +1(604) 822 7815 22 Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Stems for Nonproprietary Drug Names
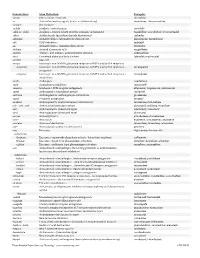
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub
US 20200187851A1TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No .: US 2020/0187851 A1 Offenbacher et al. (43 ) Pub . Date : Jun . 18 , 2020 ( 54 ) PERIODONTAL DISEASE STRATIFICATION (52 ) U.S. CI. AND USES THEREOF CPC A61B 5/4552 (2013.01 ) ; G16H 20/10 ( 71) Applicant: The University of North Carolina at ( 2018.01) ; A61B 5/7275 ( 2013.01) ; A61B Chapel Hill , Chapel Hill , NC (US ) 5/7264 ( 2013.01 ) ( 72 ) Inventors: Steven Offenbacher, Chapel Hill , NC (US ) ; Thiago Morelli , Durham , NC ( 57 ) ABSTRACT (US ) ; Kevin Lee Moss, Graham , NC ( US ) ; James Douglas Beck , Chapel Described herein are methods of classifying periodontal Hill , NC (US ) patients and individual teeth . For example , disclosed is a method of diagnosing periodontal disease and / or risk of ( 21) Appl. No .: 16 /713,874 tooth loss in a subject that involves classifying teeth into one of 7 classes of periodontal disease. The method can include ( 22 ) Filed : Dec. 13 , 2019 the step of performing a dental examination on a patient and Related U.S. Application Data determining a periodontal profile class ( PPC ) . The method can further include the step of determining for each tooth a ( 60 ) Provisional application No.62 / 780,675 , filed on Dec. Tooth Profile Class ( TPC ) . The PPC and TPC can be used 17 , 2018 together to generate a composite risk score for an individual, which is referred to herein as the Index of Periodontal Risk Publication Classification ( IPR ) . In some embodiments , each stage of the disclosed (51 ) Int. Cl. PPC system is characterized by unique single nucleotide A61B 5/00 ( 2006.01 ) polymorphisms (SNPs ) associated with unique pathways , G16H 20/10 ( 2006.01 ) identifying unique druggable targets for each stage . -
Downloaded from Survive Nursing | Survivenursing.Com V20110426
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol -

Datasheet Inhibitors / Agonists / Screening Libraries a DRUG SCREENING EXPERT
Datasheet Inhibitors / Agonists / Screening Libraries A DRUG SCREENING EXPERT Product Name : Prinaberel Catalog Number : TQ0149 CAS Number : 524684-52-4 Molecular Formula : C15H10FNO3 Molecular Weight : 271.24 Description: Prinaberel (ERB-041) is an effective and selective ERβ agonist and >200-fold selective for ERβ. Storage: 2 years -80°C in solvent; 3 years -20°C powder; Solubility DMSO ≥38mg/mL(140.1 mM) ( < 1 mg/ml refers to the product slightly soluble or insoluble ) In vitro Activity Treatment with ERβ selective estrogen agonists liquiritigenin and ERB-041 reduced the ability to invade a reconstituted basement membrane and to migrate in response to the cellular stimulus [1]. Pretreatment the PMs with ERB-041 resulted in significant inhibition of LPS-induced iNOS expression and NF-kappaB activation by preventing its nuclear translocation [3]. In vivo Activity Tumor numbers and volume were reduced by 60% and 84%, respectively, in the Erb-041-treated group as compared with UVB (alone) control. This inhibition in tumorigenesis was accompanied by a decrease in proliferating cell nuclear antigen (PCNA), cyclin D1, VEGF, and CD31, and an increase in apoptosis [2]. Reference 1. Hinsche O, et al. Estrogen receptor β selective agonists reduce invasiveness of triple-negative breast cancer cells. Int J Oncol. 2015 Feb;46(2):878-84. 2. Chaudhary SC, et al. Erb-041, an estrogen receptor-β agonist, inhibits skin photocarcinogenesis in SKH-1 hairless mice by downregulating the WNT signaling pathway. Cancer Prev Res (Phila). 2014 Feb;7(2):186-98. 3. Xiu-li W, et al. ERB-041, a selective ER beta agonist, inhibits iNOS production in LPS-activated peritoneal macrophages of endometriosis via suppression of NF-kappaB activation. -

The Concise Guide to PHARMACOLOGY 2015/16
Edinburgh Research Explorer The Concise Guide to PHARMACOLOGY 2015/16 Citation for published version: CGTP Collaborators, Alexander, SP, Cidlowski, JA, Kelly, E, Marrion, N, Peters, JA, Benson, HE, Faccenda, E, Pawson, AJ, Sharman, JL, Southan, C & Davies, JA 2015, 'The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors', British Journal of Pharmacology, vol. 172, no. 24, pp. 5956-5978. https://doi.org/10.1111/bph.13352 Digital Object Identifier (DOI): 10.1111/bph.13352 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 30. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. British Journal of Pharmacology (2015) 172, 5956–5978 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: Nuclear hormone -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

A Abacavir Abacavirum Abakaviiri Abagovomab Abagovomabum
A abacavir abacavirum abakaviiri abagovomab abagovomabum abagovomabi abamectin abamectinum abamektiini abametapir abametapirum abametapiiri abanoquil abanoquilum abanokiili abaperidone abaperidonum abaperidoni abarelix abarelixum abareliksi abatacept abataceptum abatasepti abciximab abciximabum absiksimabi abecarnil abecarnilum abekarniili abediterol abediterolum abediteroli abetimus abetimusum abetimuusi abexinostat abexinostatum abeksinostaatti abicipar pegol abiciparum pegolum abisipaaripegoli abiraterone abirateronum abirateroni abitesartan abitesartanum abitesartaani ablukast ablukastum ablukasti abrilumab abrilumabum abrilumabi abrineurin abrineurinum abrineuriini abunidazol abunidazolum abunidatsoli acadesine acadesinum akadesiini acamprosate acamprosatum akamprosaatti acarbose acarbosum akarboosi acebrochol acebrocholum asebrokoli aceburic acid acidum aceburicum asebuurihappo acebutolol acebutololum asebutololi acecainide acecainidum asekainidi acecarbromal acecarbromalum asekarbromaali aceclidine aceclidinum aseklidiini aceclofenac aceclofenacum aseklofenaakki acedapsone acedapsonum asedapsoni acediasulfone sodium acediasulfonum natricum asediasulfoninatrium acefluranol acefluranolum asefluranoli acefurtiamine acefurtiaminum asefurtiamiini acefylline clofibrol acefyllinum clofibrolum asefylliiniklofibroli acefylline piperazine acefyllinum piperazinum asefylliinipiperatsiini aceglatone aceglatonum aseglatoni aceglutamide aceglutamidum aseglutamidi acemannan acemannanum asemannaani acemetacin acemetacinum asemetasiini aceneuramic -

Nuclear Hormone Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology (2017) 174, S208–S224 THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Nuclear hormone receptors Stephen PH Alexander1, John A Cidlowski2, Eamonn Kelly3, Neil V Marrion3, John A Peters4, Elena Faccenda5, Simon D Harding5,AdamJPawson5, Joanna L Sharman5, Christopher Southan5, Jamie A Davies5 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, Research Triangle Park, NC 27709, USA 3School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 4Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 5Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2017/18 provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to an open access knowledgebase of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. Although the Concise Guide represents approximately 400 pages, the material presented is substantially reduced compared to information and links presented on the website. It provides a permanent, citable, point-in-time record that will survive database updates. The full contents of this section can be found at http://onlinelibrary.wiley.com/doi/10.1111/bph.13880/full. Nuclear hormone receptors are one of the eight major pharmacological targets into which the Guide is divided, with the others being: G protein-coupled receptors, ligand-gated ion channels, voltage-gated ion channels, other ion channels, catalytic receptors, enzymes and transporters. -

WO 2009/143297 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 26 November 2009 (26.11.2009) WO 2009/143297 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every AOlN 57/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) International Application Number: CA, CH, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, PCT/US2009/044746 EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, (22) International Filing Date: HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, 20 May 2009 (20.05.2009) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, (25) Filing Language: English NZ, OM, PG, PH, PL, PT, RO, RS, RU, SC, SD, SE, SG, (26) Publication Language: English SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/054,765 20 May 2008 (20.05.2008) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): NEU- GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, ROGESX, INC. [US/US]; 2215 Bridgepointe Parkway, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, Suite 200, San Mateo, CA 94404 (US). -

(12) United States Patent (10) Patent No.: US 8,735,376 B2 Muhammad Et Al
US008735376B2 (12) United States Patent (10) Patent No.: US 8,735,376 B2 Muhammad et al. (45) Date of Patent: May 27, 2014 (54) CARBONATE PRODRUGS AND METHODS 2004/0058946 A1 3/2004 Buchwald et al. OF USING THE SAME 2004/O186081 A1 9, 2004 Slusher et al. 2005.0020576 A1 1/2005 Zhang et al. 2005, 0026850 A1 2/2005 Robinson et al. (75) Inventors: Naweed Muhammad, Fremont, CA 2005/0026879 A1 2/2005 Robinson et al. (US); Keith R. Bley, Mountain View, 2005, 0080260 A1 4/2005 Mills et al. CA (US) 2005, 01476.68 A1 7/2005 Bertelsen et al. 2006.0089383 A1 4/2006 Le Bourdonnec et al. Assignee: 2006, O116422 A1 6/2006 De Groot et al. (73) Acorda Therapeutics, Inc., Ardsley, NY 2007/0042999 A1 2/2007 West et al. (US) 2008/0318905 A1 12/2008 Muhammad et al. 2011/0212926 A1 9/2011 Muhammad et al. (*) Notice: Subject to any disclaimer, the term of this 2011/0263545 A1 10, 2011 Muhammad et al. patent is extended or adjusted under 35 U.S.C. 154(b) by 328 days. FOREIGN PATENT DOCUMENTS Appl. No.: 12/993,089 WO WO-0008033 A1 2, 2000 (21) WO WO-O 1/52852 A1 T 2001 WO WO-2005, O77394 A1 8, 2005 (22) PCT Fled: May 20, 2009 WO WO-2006/O14282 A2 2, 2006 WO WO-2009/143295 A1 11, 2009 (86) PCT NO PCT/US2O09/044746 WO WO-2009/143297 A1 11, 2009 S371 (c)(1), WO WO-2009/143299 A1 11, 2009 (2), (4) Date: Apr. -

Berrchem Company Ltd API List
Berrchem Company Ltd API List 136470-78-5 Aba cavir 154229-19-3 Abiraterone 226954-04-7 AC-5216 57960-19-7 Ace qui nocyl 6622-91-9 4-(A cetic A cid) Pyridine hydr ochloride 5 -Acetylamino-2-a min o-4-picoli ne 93273-63-3 4-Acetyl-2-bromobenz onitrile 49 669 -13-8 2-A cetyl-6-bromopyridine 561 72-71-5 1-Acetyl-cycl opropane carboxylic aci d 57 756 -36-2 4-Acetyl -2,3,4, 5-tetrahydr o-1H-1, 4-benz odiaz epine 550 79-83-9 Acitretin 55077-30-0 Aclatonium napadisilate 10523-68-9 2-A dama ntanami ne hydrochlori de 10 6941 -25-7 Adefovir 1991-10-1 aldrich 80863-62-3 A LITAME 315-30-0 Allopurinol 850649-62-6 Alogliptin benzoat e 25526-93-6 Alovudi ne 8 50-52-2 ALT RENOGEST 645-05-6 Altretami ne 177036-94-1 AMBRISENTAN 61618-27-7 Amfe nac sodi um 773 7-17-9 Ami noacetone hydr ochl oride 2498 -50-2 4-A minobenzami dine Dihydrochlori de 3 7141-01-8 1-A minobenzene -3,4, 5-tricarboxylic acid 23031-78-9 3-A mino -1, 2- benzisothiazole 7644-4-4 2-Ami no -4’-bromoa cetophenone HCl 20776-51-6 2-a mino -3-bromobenzoic aci d 135776-98-6 (2-A mino -4-bro mo -phenyl)-phenyl-metha none 2-Amin o-4’-chlor oacetophe none HCl salt 89604-91-1 7-Ami no -3-Chl oro Cephalos porani c Acid 5399 4-69-7 7-Amin o-3-Chlor o Cephal ospora nic Acid 112 885 -41-3 4-A mino -5-chloro-2-etho xy-N-((4 -(4-fluor obe nzyl)-2-morpholinyl)methyl)benza mide 100371 0-83-5 2-Amin o-5-chlor o-4-fluor o-3-hydr oxypyridi ne 14394-56-0 6-A mino -4-Chl oro -5-M ethylpyrimidi ne 51564 -92-2 2-A min o-6-chloro -4-pi coline 58483-95-7 5-AMI NO-2-CHLO RO PYRIDI NE-4-CA RBOXY LIC ACID 214353-17-0