Combinatorial Treatments of Tamoxifen with Sm6met, a Selective Estrogen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting Fibrosis in the Duchenne Muscular Dystrophy Mice Model: an Uphill Battle
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Targeting fibrosis in the Duchenne Muscular Dystrophy mice model: an uphill battle 2 Marine Theret1#, Marcela Low1#, Lucas Rempel1, Fang Fang Li1, Lin Wei Tung1, Osvaldo 3 Contreras3,4, Chih-Kai Chang1, Andrew Wu1, Hesham Soliman1,2, Fabio M.V. Rossi1 4 1School of Biomedical Engineering and the Biomedical Research Centre, Department of Medical 5 Genetics, 2222 Health Sciences Mall, Vancouver, BC, V6T 1Z3, Canada 6 2Department of Pharmacology and Toxicology, Faculty of Pharmaceutical Sciences, Minia 7 University, Minia, Egypt 8 3Developmental and Stem Cell Biology Division, Victor Chang Cardiac Research Institute, 9 Darlinghurst, NSW, 2010, Australia 10 4Departamento de Biología Celular y Molecular and Center for Aging and Regeneration (CARE- 11 ChileUC), Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, 8331150 12 Santiago, Chile 13 # Denotes Co-first authorship 14 15 Keywords: drug screening, fibro/adipogenic progenitors, fibrosis, repair, skeletal muscle. 16 Correspondence to: 17 Marine Theret 18 School of Biomedical Engineering and the Biomedical Research Centre 19 University of British Columbia 20 2222 Health Sciences Mall, Vancouver, British Columbia 21 Tel: +1(604) 822 0441 fax: +1(604) 822 7815 22 Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Mcfeely Washington 0250E 19
© Copyright 2019 Savannah Jane Kerr McFeely Clinical Significance and Regulatory Framework for the Evaluation of Organic Anion Transporting Polypeptide 1B-Based Drug-Drug Interactions Savannah Jane Kerr McFeely A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2019 Reading Committee: Isabelle Ragueneau-Majlessi, Chair Jashvant Unadkat Bhagwat Prasad Program Authorized to Offer Degree: Pharmaceutics University of Washington Abstract Clinical Significance and Regulatory Framework for the Evaluation of Organic Anion Transporting Polypeptide 1B-Based Drug-Drug Interactions Savannah Jane Kerr McFeely Chair of the Supervisory Committee: Dr. Isabelle Ragueneau-Majlessi Department of Pharmaceutics This dissertation research aims to conduct a comprehensive analysis of the clinical role of organic anion transporting polypeptides (OATPs) 1B1 and 1B3 and the regulatory approach for their evaluation. The work included here has identified the most relevant clinical substrates and inhibitors. Additionally, the contributing factors in the variability of in vitro inhibition constants as well as real-world implications for OATP1B1/1B3 drug-drug interactions are discussed. In Chapter 2, six compounds were identified as potential clinical markers of OATP1B1/1B3 activity through the use of a novel indexing system. These drugs were identified from a list of 34 clinical substrates identified from a thorough analysis of the available in vitro and in vivo data. These findings also suggest that the risk for comedication interactions involve drugs from multiple therapeutic areas showing a reliance on hepatic uptake via the OATP transporters. Chapters 3 seeks to better understand the variability of in vitro inhibition data. By analyzing available OATP1B1/1B3 IC50 values, the primary contributing factors to in vitro variability were identified as the cell system used and inclusion of a preincubation with the inhibitor. -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub
US 20200187851A1TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No .: US 2020/0187851 A1 Offenbacher et al. (43 ) Pub . Date : Jun . 18 , 2020 ( 54 ) PERIODONTAL DISEASE STRATIFICATION (52 ) U.S. CI. AND USES THEREOF CPC A61B 5/4552 (2013.01 ) ; G16H 20/10 ( 71) Applicant: The University of North Carolina at ( 2018.01) ; A61B 5/7275 ( 2013.01) ; A61B Chapel Hill , Chapel Hill , NC (US ) 5/7264 ( 2013.01 ) ( 72 ) Inventors: Steven Offenbacher, Chapel Hill , NC (US ) ; Thiago Morelli , Durham , NC ( 57 ) ABSTRACT (US ) ; Kevin Lee Moss, Graham , NC ( US ) ; James Douglas Beck , Chapel Described herein are methods of classifying periodontal Hill , NC (US ) patients and individual teeth . For example , disclosed is a method of diagnosing periodontal disease and / or risk of ( 21) Appl. No .: 16 /713,874 tooth loss in a subject that involves classifying teeth into one of 7 classes of periodontal disease. The method can include ( 22 ) Filed : Dec. 13 , 2019 the step of performing a dental examination on a patient and Related U.S. Application Data determining a periodontal profile class ( PPC ) . The method can further include the step of determining for each tooth a ( 60 ) Provisional application No.62 / 780,675 , filed on Dec. Tooth Profile Class ( TPC ) . The PPC and TPC can be used 17 , 2018 together to generate a composite risk score for an individual, which is referred to herein as the Index of Periodontal Risk Publication Classification ( IPR ) . In some embodiments , each stage of the disclosed (51 ) Int. Cl. PPC system is characterized by unique single nucleotide A61B 5/00 ( 2006.01 ) polymorphisms (SNPs ) associated with unique pathways , G16H 20/10 ( 2006.01 ) identifying unique druggable targets for each stage . -

Emtree Terms Changed in January 2017
Emtree Terms Added and Changed (January 2017) This is an overview of new terms added and changes made in the first Emtree release in 2017. Overall, Emtree has grown by 782 preferred terms (269 drug terms and 513 non-drug terms) compared with the previous version released in September 2016. In total Emtree now counts 75,488 preferred terms. Because the terms added include replacements for existing preferred terms (which become synonyms of the new terms) as well as completely new concepts, the number of terms added exceeds the net growth in Emtree. Other changes could include the merging of two or more existing preferred terms into a single concept. The terms added and changed are summarized below and specified in detail on the following pages. Emtree Terms Added in January 2017 930 new terms (including 148 replacement terms and promoted synonyms) have been added to Emtree as preferred terms in version January 2017 (compared to September 2016): 308 drug terms (terms assigned to the Chemicals and Drugs facet). 622 non-drug terms (terms not assigned as Chemicals and Drugs). The new terms (including the replacement terms and the promoted synonyms) are listed as Terms Added on the following pages. Note that many of these terms will have been indexed prior to 2017 (typically as candidate terms), sometimes for several years, before they were added to Emtree. Emtree Terms Changed in January 2017 148 terms (39 drug terms and 109 non-drug terms) from Emtree September 2016 have been replaced by 148 different terms in January 2017 (39 drug terms and 109 non-drug terms). -
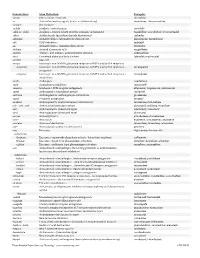
Downloaded from Survive Nursing | Survivenursing.Com V20110426
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol -

Datasheet Inhibitors / Agonists / Screening Libraries a DRUG SCREENING EXPERT
Datasheet Inhibitors / Agonists / Screening Libraries A DRUG SCREENING EXPERT Product Name : Prinaberel Catalog Number : TQ0149 CAS Number : 524684-52-4 Molecular Formula : C15H10FNO3 Molecular Weight : 271.24 Description: Prinaberel (ERB-041) is an effective and selective ERβ agonist and >200-fold selective for ERβ. Storage: 2 years -80°C in solvent; 3 years -20°C powder; Solubility DMSO ≥38mg/mL(140.1 mM) ( < 1 mg/ml refers to the product slightly soluble or insoluble ) In vitro Activity Treatment with ERβ selective estrogen agonists liquiritigenin and ERB-041 reduced the ability to invade a reconstituted basement membrane and to migrate in response to the cellular stimulus [1]. Pretreatment the PMs with ERB-041 resulted in significant inhibition of LPS-induced iNOS expression and NF-kappaB activation by preventing its nuclear translocation [3]. In vivo Activity Tumor numbers and volume were reduced by 60% and 84%, respectively, in the Erb-041-treated group as compared with UVB (alone) control. This inhibition in tumorigenesis was accompanied by a decrease in proliferating cell nuclear antigen (PCNA), cyclin D1, VEGF, and CD31, and an increase in apoptosis [2]. Reference 1. Hinsche O, et al. Estrogen receptor β selective agonists reduce invasiveness of triple-negative breast cancer cells. Int J Oncol. 2015 Feb;46(2):878-84. 2. Chaudhary SC, et al. Erb-041, an estrogen receptor-β agonist, inhibits skin photocarcinogenesis in SKH-1 hairless mice by downregulating the WNT signaling pathway. Cancer Prev Res (Phila). 2014 Feb;7(2):186-98. 3. Xiu-li W, et al. ERB-041, a selective ER beta agonist, inhibits iNOS production in LPS-activated peritoneal macrophages of endometriosis via suppression of NF-kappaB activation. -

Oncompass Report NÉV Anonymous a Realtime Oncology Molecular Treatment Calculator Számításaival
FIGYELMEZTETÉS Ezt a tájékoztatót csak a kezelőorvos használhatja és értelmezheti. Az orvos mérlegelheti, vagy figyelmen kívül hagyhatja a jelentés által nyújtott információkat. Az Oncompass Riport információt szolgáltat a tumorok és a molekuláris profil közti összefüggésekről a tudományos irodalom felhasználásával. Az ONCOMPASS Medicine a szakirodalom tartalmáért felelősséget nem vállal. A feltüntetett gyógyszerek az adott tumortípusban lehetnek törzskönyvezettek és/vagy finanszírozottak, annak viszonylatában, hogy a riportot melyik országban használják. AZONOSÍTÓ Oncompass Report NÉV Anonymous A Realtime Oncology Molecular Treatment Calculator számításaival BETEG ADATAI OncompassTM ID: Primer daganat lokalizációja: breast Név: Szövettani típus: invasive carcinoma NST Születési dátum: 1959 Metasztázis lokalizációja: bone, lymph node SZAKÉRTŐK Molekuláris Farmakológus: Dr. Peták István Genetikai Tanácsadó: Déri Júlia, MSc Molekuláris Biológus: Várkondi Edit, PhD Konzulens Orvos: Dr. Pajkos Gábor Szakértő: Dr. Szuszán Marianna Betegút Koordinátor: Czető Réka Info-bionikus Mérnök: Tihanyi Dóra, MSc Molekuláris Bionikus Mérnök: Dirner Anna, MSc PATOLÓGIAI ÉS MOLEKULÁRIS DIAGNOSZTIKAI VIZSGÁLATOK Mintaazonosító: XXX (szövettani minta) Minta eredete: nyirokcsomó metasztázis Tumorarány: 50% Tumortípus: invazív ductális emlő carcinoma Elvégzett vizsgálatok: IHC - PD-L1 normál expresszió MSI - MSS TMB - magas: 6,44 mut/Mb NGS - 591 gén Korábbi molekuláris vizsgálatok eredménye: IHC - ER, PR, HER2 normál expresszió IHC - PD-L1 overexpresszió (>1%) -

The Concise Guide to PHARMACOLOGY 2015/16
Edinburgh Research Explorer The Concise Guide to PHARMACOLOGY 2015/16 Citation for published version: CGTP Collaborators, Alexander, SP, Cidlowski, JA, Kelly, E, Marrion, N, Peters, JA, Benson, HE, Faccenda, E, Pawson, AJ, Sharman, JL, Southan, C & Davies, JA 2015, 'The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors', British Journal of Pharmacology, vol. 172, no. 24, pp. 5956-5978. https://doi.org/10.1111/bph.13352 Digital Object Identifier (DOI): 10.1111/bph.13352 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 30. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. British Journal of Pharmacology (2015) 172, 5956–5978 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: Nuclear hormone -

Orally Active Compound Library (96-Well)
• Bioactive Molecules • Building Blocks • Intermediates www.ChemScene.com Orally Active Compound Library (96-well) Product Details: Catalog Number: CS-L061 Formulation: A collection of 2244 orally active compounds supplied as pre-dissolved Solutions or Solid Container: 96- or 384-well Plate with Peelable Foil Seal; 96-well Format Sample Storage Tube With Screw Cap and Optional 2D Barcode Storage: -80°C Shipping: Blue ice Packaging: Inert gas Plate layout: CS-L061-1 1 2 3 4 5 6 7 8 9 10 11 12 a Empty Vonoprazan Mivebresib AZD0156 BAY-876 Tomivosertib PQR620 NPS-2143 R547 PAP-1 Delpazolid Empty Varenicline b Empty Varenicline (Hydrochlorid Varenicline A-205804 CI-1044 KW-8232 GSK583 Quiflapon MRK-016 Diroximel Empty e) (Tartrate) fumarate SHP099 c Empty A 922500 SHP099 (hydrochlorid Nevanimibe Pactimibe Pactimibe GNF-6231 MLi-2 KDM5-IN-1 PACMA 31 Empty e) hydrochloride (sulfate) Rilmenidine d Empty Cadazolid Treprostinil GSK3179106 Esaxerenone (hemifumarat Rilmenidine Fisogatinib BP-1-102 Avadomide Aprepitant Empty e) (phosphate) Cl-amidine AZD5153 (6- e Empty Maropitant Abscisic acid (hydrochlorid BGG463 WNK463 Ticagrelor Axitinib Hydroxy-2- CGS 15943 Pipamperone Empty e) naphthoic Setogepram Teglarinad f Empty Y-27632 GSK2193874 Lanabecestat CXD101 Ritlecitinib YL0919 PCC0208009 (sodium salt) (chloride) Futibatinib Empty TAK-659 g Empty NCB-0846 GS-444217 (hydrochlorid Navitoclax ABX464 Zibotentan Simurosertib (R)- CEP-40783 MK-8617 Empty e) Simurosertib Choline JNJ- h Empty CCT251236 (bitartrate) Sarcosine Brensocatib AS-605240 PF-06840003 -

Effect of Estrogen on Host-Pathogen Interactions in Ex Vivo and in Vitro Models of the Inflammatory Phase
Effect of Estrogen on Host-Pathogen Interactions in ex vivo and in vitro Models of the Inflammatory Phase of Age-Related Impaired Healing A thesis submitted in partial fulfilment of the requirements of Manchester Metropolitan University for the degree of Doctor of Philosophy Mohamed El Mohtadi Department of Life Sciences Manchester Metropolitan University 2019 Table of Contents List of Abbreviations………………………………………………………………………………………VI List of Figures………………………………………………………………………….……………………..X List of Tables………………………………………………………………………………………………….XIII Abstract…………………………………………………………………………………………………………XIV Declaration and Copyright Statements…………………………………………………………..XVI The Author…………………………………………………………………………………………………….XVII Acknowledgments………………………………………………………………………………………...XVIII Dedication……………………………………………………………………………………………………..XX : Introduction .................................................................................... 1 1.1 Acute Wound Healing ..................................................................................... 2 1.1.1 Haemostasis ................................................................................................................... 4 1.1.2 Inflammatory Phase ....................................................................................................... 5 1.1.3 Proliferative Phase ......................................................................................................... 8 ECM Formation .................................................................................................... -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.