University of Oklahoma Graduate College
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

REVIEW Electric Fish: New Insights Into Conserved Processes of Adult Tissue Regeneration
2478 The Journal of Experimental Biology 216, 2478-2486 © 2013. Published by The Company of Biologists Ltd doi:10.1242/jeb.082396 REVIEW Electric fish: new insights into conserved processes of adult tissue regeneration Graciela A. Unguez Department of Biology, New Mexico State University, Las Cruces, NM 88003, USA [email protected] Summary Biology is replete with examples of regeneration, the process that allows animals to replace or repair cells, tissues and organs. As on land, vertebrates in aquatic environments experience the occurrence of injury with varying frequency and to different degrees. Studies demonstrate that ray-finned fishes possess a very high capacity to regenerate different tissues and organs when they are adults. Among fishes that exhibit robust regenerative capacities are the neotropical electric fishes of South America (Teleostei: Gymnotiformes). Specifically, adult gymnotiform electric fishes can regenerate injured brain and spinal cord tissues and restore amputated body parts repeatedly. We have begun to identify some aspects of the cellular and molecular mechanisms of tail regeneration in the weakly electric fish Sternopygus macrurus (long-tailed knifefish) with a focus on regeneration of skeletal muscle and the muscle-derived electric organ. Application of in vivo microinjection techniques and generation of myogenic stem cell markers are beginning to overcome some of the challenges owing to the limitations of working with non-genetic animal models with extensive regenerative capacity. This review highlights some aspects of tail regeneration in S. macrurus and discusses the advantages of using gymnotiform electric fishes to investigate the cellular and molecular mechanisms that produce new cells during regeneration in adult vertebrates. -

Convergent Evolution of Mechanically Optimal Locomotion in Aquatic Invertebrates and Vertebrates
RESEARCH ARTICLE Convergent Evolution of Mechanically Optimal Locomotion in Aquatic Invertebrates and Vertebrates Rahul Bale1, Izaak D. Neveln2, Amneet Pal Singh Bhalla1, Malcolm A. MacIver1,2,3☯*, Neelesh A. Patankar1☯* 1 Department of Mechanical Engineering, Northwestern University, Evanston, Illinois, United States of America, 2 Department of Biomedical Engineering, Northwestern University, Evanston, Illinois, United States of America, 3 Department of Neurobiology, Northwestern University, Evanston, Illinois, United States of America ☯ These authors contributed equally to this work. * [email protected] (NAP); [email protected] (MAM) Abstract OPEN ACCESS Examples of animals evolving similar traits despite the absence of that trait in the last com- Citation: Bale R, Neveln ID, Bhalla APS, MacIver MA, Patankar NA (2015) Convergent Evolution of mon ancestor, such as the wing and camera-type lens eye in vertebrates and invertebrates, Mechanically Optimal Locomotion in Aquatic are called cases of convergent evolution. Instances of convergent evolution of locomotory Invertebrates and Vertebrates. PLoS Biol 13(4): patterns that quantitatively agree with the mechanically optimal solution are very rare. Here, e1002123. doi:10.1371/journal.pbio.1002123 we show that, with respect to a very diverse group of aquatic animals, a mechanically opti- Academic Editor: Anders Hedenström, Lund mal method of swimming with elongated fins has evolved independently at least eight times University, SWEDEN in both vertebrate and invertebrate swimmers across three different phyla. Specifically, if we Received: September 29, 2014 take the length of an undulation along an animal’s fin during swimming and divide it by the Accepted: March 6, 2015 mean amplitude of undulations along the fin length, the result is consistently around twenty. -

Advances in the Study of Behavior, Volume 31.Pdf
Advances in THE STUDY OF BEHAVIOR VOLUME 31 Advances in THE STUDY OF BEHAVIOR Edited by PETER J. B. S LATER JAY S. ROSENBLATT CHARLES T. S NOWDON TIMOTHY J. R OPER Advances in THE STUDY OF BEHAVIOR Edited by PETER J. B. S LATER School of Biology University of St. Andrews Fife, United Kingdom JAY S. ROSENBLATT Institute of Animal Behavior Rutgers University Newark, New Jersey CHARLES T. S NOWDON Department of Psychology University of Wisconsin Madison, Wisconsin TIMOTHY J. R OPER School of Biological Sciences University of Sussex Sussex, United Kingdom VOLUME 31 San Diego San Francisco New York Boston London Sydney Tokyo This book is printed on acid-free paper. ∞ Copyright C 2002 by ACADEMIC PRESS All Rights Reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the Publisher. The appearance of the code at the bottom of the first page of a chapter in this book indicates the Publisher’s consent that copies of the chapter may be made for personal or internal use of specific clients. This consent is given on the condition, however, that the copier pay the stated per copy fee through the Copyright Clearance Center, Inc. (222 Rosewood Drive, Danvers, Massachusetts 01923), for copying beyond that permitted by Sections 107 or 108 of the U.S. Copyright Law. This consent does not extend to other kinds of copying, such as copying for general distribution, for advertising or promotional purposes, for creating new collective works, or for resale. -
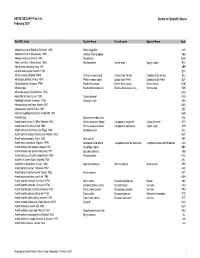
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Diversity and Phylogeny of Neotropical Electric Fishes (Gymnotiformes)
Name /sv04/24236_u11 12/08/04 03:33PM Plate # 0-Composite pg 360 # 1 13 Diversity and Phylogeny of Neotropical Electric Fishes (Gymnotiformes) James S. Albert and William G.R. Crampton 1. Introduction to Gymnotiform Diversity The evolutionary radiations of Neotropical electric fishes (Gymnotiformes) pro- vide unique materials for studies on the evolution of specialized sensory systems and the diversification of animals species in tropical ecosystems (Hopkins and Heiligenberg 1978; Heiligenberg 1980; Heiligenberg and Bastian 1986; Moller 1995a; Crampton 1998a; Stoddard 1999; Albert 2001, 2002). The teleost order Gymnotiformes is a clade of ostariophysan fishes most closely related to cat- fishes (Siluriformes), with which they share the presence of a passive electro- sensory system (Fink and Fink 1981, 1996; Finger 1986). Gymnotiformes also possess a combined electrogenic–electroreceptive system that is employed for both active electrolocation, the detection of nearby objects that distort the self- generated electric field, and also electrocommunication, the signaling of identity or behavioral states and intentions to other fishes (Carr and Maler 1986). Active electroreception allows gymnotiforms to communicate, navigate, forage, and ori- ent themselves relative to the substrate at night and in dark, sediment-laden waters, and contributes to their ecological success in Neotropical aquatic eco- systems (Crampton and Albert 2005). The species-specific electric signals of gymnotiform fishes allow investigations of behavior and ecology that are simply unavailable in other groups. Because these signals are used in both navigation and mate recognition (i.e., prezygotic reproductive isolation) they play central roles in the evolutionary diversification and ecological specialization of species, as well as the accumulation of species into local and regional assem- blages. -

Nonlinear Processing of Sensory Interference
NONLINEAR PROCESSING OF SENSORY INTERFERENCE DRIVES SOCIAL BEHAVIOR IN WEAKLY ELECTRIC FISH by Manu S. Madhav A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy. Baltimore, Maryland March, 2014 c Manu S. Madhav 2014 All rights reserved Abstract Sensory systems in animals and robots can receive information from the environ- ment via modulations of sensory signals. Environmental features and conspecifics generate these modulations, and effective computational algorithms in sensory sys- tems can demodulate signals to perceive the features. However, interaction of mul- tiple modulations can themselves generate emergent modulations, which can encode relative information between environmental features. These emergent modulations create sensory interference which can be detrimental or beneficial to sensing. Or- ganisms that probe their environment using autogenous sensory signals are prone to such interference from other nearby individuals in the same social group. Some animals have evolved behaviors that allow them to retain a high level of sensory performance in such a social context. For example, the weakly electric glass knife- fish, Eigenmannia virescens, produces a pseudo-sinusoidal oscillating electric signal used for electrolocation and social communication. In my thesis, I explore how the electrosensory system of weakly electric fish use nonlinear processing to respond to interference from conspecifics. ii ABSTRACT In Eigenmannia and other wave-type electric fishes, the interaction between two individuals produces first-order modulations termed beats, and interaction between three or more individuals produce \beats of beats", termed envelopes. In this thesis, I provide an analytical basis for the emergence of beats and envelopes in sensory systems. -

Behavioral and Neural Aspects of the Spatial Processing of Conspecific Signals in the Electrosensory System
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 20 March 2019 Spatial Processing in the Electrosensory System Behavioral and neural aspects of the spatial processing of conspecific signals in the electrosensory system Oak E Milam*, Keshav L Ramachandra* and Gary Marsat Biology Department, West Virginia University * contributed equally to this manuscript Corresponding author: Gary Marsat [email protected] 53 Campus Dr, WVU Biology Dept, Morgantown, WV, 26506 Acknowledgements: We thank Len Maler, Andrew Dacks and Frederico Predaja for helpful comments on the manuscript. 1 © 2019 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 20 March 2019 Spatial Processing in the Electrosensory System Abstract Localizing the source of a signal is often as important as deciphering the signal’s message. Localization mechanisms must cope with the challenges of representing the spatial information of weak, noisy signals. Comparing these strategies across modalities and model systems allows a broader understanding of the general principles shaping spatial processing. In this review we focus on the electrosensory system of knifefish and provide an overview of our current understanding of spatial processing in this system, in particular, localization of conspecific signals. We argue that many mechanisms observed in other sensory systems, such as the visual or auditory systems, have comparable implementations in the electrosensory system. Our review therefore describes a field of research with unique opportunities to provide new insights into the principles underlying spatial processing. Keywords: spatial information; localization of conspecifics; neural mechanisms; sensory processing; sensing behavior. 2 Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 20 March 2019 Spatial Processing in the Electrosensory System Introduction The role of sensory systems is to capture information about the environment. -

Spooky Interaction at a Distance in Cave and Surface Dwelling Electric fishes Eric S
bioRxiv preprint doi: https://doi.org/10.1101/747154; this version posted May 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. submitted to 1 Spooky interaction at a distance in cave and surface dwelling electric fishes Eric S. Fortune1;∗, Nicole Andanar1, Manu Madhav2, Ravi Jayakumar2, Noah J. Cowan2, Maria Elina Bichuette3, and Daphne Soares1 1New Jersey Institute of Technology, Biological Sciences, Newark, 07102, USA 2Johns Hopkins University, Whiting School of Engineering, Baltimore, 21218, USA 3Universidade Federal de Sao˜ Carlos, Departamento de Ecologia e Biologia Evolutiva, Sao˜ Carlos, 13565-905, Brasil Correspondence*: Eric Fortune [email protected] 2 ABSTRACT 3 Glass knifefish (Eigenmannia) are a group of weakly electric fishes found throughout the Amazon 4 basin. We made recordings of the electric fields of two populations of freely behaving Eigenmannia 5 in their natural habitats: a troglobitic population of blind cavefish (Eigenmannia vicentespelaea) 6 and a nearby epigean (surface) population (Eigenmannia trilineata). These recordings were 7 made using a grid of electrodes to determine the movements of individual fish in relation to their 8 electrosensory behaviors. The strengths of electric discharges in cavefish were larger than in 9 surface fish, which may be a correlate of increased reliance on electrosensory perception and 10 larger size. Both movement and social signals were found to affect the electrosensory signaling 11 of individual Eigenmannia. Surface fish were recorded while feeding at night and did not show 12 evidence of territoriality. In contrast, cavefish appeared to maintain territories. -

Regional Biosecurity Plan for Micronesia and Hawaii Volume II
Regional Biosecurity Plan for Micronesia and Hawaii Volume II Prepared by: University of Guam and the Secretariat of the Pacific Community 2014 This plan was prepared in conjunction with representatives from various countries at various levels including federal/national, state/territory/commonwealth, industry, and non-governmental organizations and was generously funded and supported by the Commander, Navy Installations Command (CNIC) and Headquarters, Marine Corps. MBP PHASE 1 EXECUTIVE SUMMARY NISC Executive Summary Prepared by the National Invasive Species Council On March 7th, 2007 the U.S. Department of Navy (DoN) issued a Notice of Intent to prepare an “Environmental Impact Statement (EIS)/Overseas Environmental Impact Statement (OEIS)” for the “Relocation of U.S. Marine Corps Forces to Guam, Enhancement of Infrastructure and Logistic Capabilities, Improvement of Pier/Waterfront Infrastructure for Transient U.S. Navy Nuclear Aircraft Carrier (CVN) at Naval Base Guam, and Placement of a U.S. Army Ballistic Missile Defense (BMD) Task Force in Guam”. This relocation effort has become known as the “build-up”. In considering some of the environmental consequences of such an undertaking, it quickly became apparent that one of the primary regional concerns of such a move was the potential for unintentional movement of invasive species to new locations in the region. Guam has already suffered the eradication of many of its native species due to the introduction of brown treesnakes and many other invasive plants, animals and pathogens cause tremendous damage to its economy and marine, freshwater and terrestrial ecosystems. DoN, in consultation and concurrence with relevant federal and territorial regulatory entities, determined that there was a need to develop a biosecurity plan to address these concerns. -

University of Oklahoma Graduate College
UNIVERSITY OF OKLAHOMA GRADUATE COLLEGE UNIONID DRIFT DISPERSAL IN SMALL RIVERS A DISSERTATION SUBMITTED TO THE GRADUATE FACULTY in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY By PASCAL IRMSCHER Norman, Oklahoma 2014 UNIONID DRIFT DISPERSAL IN SMALL RIVERS A DISSERTATION APPROVED FOR THE DEPARTMENT OF BIOLOGY BY ___________________________________ Dr. Caryn C. Vaughn, Chair ___________________________________ Dr. Jason P. Julian ___________________________________ Dr. Jeffrey F. Kelly ___________________________________ Dr. Ingo Schlupp ___________________________________ Dr. Edith C. Marsh-Matthews © Copyright by PASCAL IRMSCHER 2014 All Rights Reserved. Les rivières sont des chemins qui marchent, et qui portent où l’on veut aller. Blaise Pascal (1623-1662) FOR MY FAMILY ACKNOWLEDGEMENTS First and foremost, I would like to thank Dr. Caryn C. Vaughn for all her help and support during the last couple of years. I would not have been able to accomplish this dissertation without her, and will be forever grateful for the things I have learned as her student. I would also like to thank all members of my Ph.D. committee, Dr. Julian, Dr. Kelly, Dr. Schlupp., and Dr. Marsh-Matthews, for their support, input, and revisions of my chapters. Many thanks go to all the staff of the Oklahoma Biological Survey (in particular to Ms. Madding and Mrs. Steil) and of the University of Oklahoma Biology Department (namely Mr. Martin). The Department of Biology, its faculty, and all staff have been supportive not only in a financial and educational way, but I am even more so grateful for the wonderful people who did everything they could to help me to be able to finish my studies at the University of Oklahoma. -

Feedback Control As a Framework for Understanding Tradeoffs in Biology
Feedback Control as a Framework for Understanding Tradeoffs in Biology Noah J. Cowan1;∗, M. Mert Ankaralı1, Jonathan2 P. Dyhr, Manu S. Madhav1, Eatai Roth2, Shahin Sefati1, Simon Sponberg2, Sarah A. Stamper1, Eric S. Fortune3 and Thomas L. Daniel2 1Department of Mechanical Engineering, Johns Hopkins University, Baltimore, MD 21218, USA, 2Department of Biology, University of Washington, Seattle, WA 98195, USA, and 3Department of Biological Sciences, New Jersey Institute of Technology, Newark, NJ 07102, USA ∗Author for correspondence ([email protected]) Abstract considering a fundamental organizational feature inherent to biological systems: feedback regulation. Living systems ubiq- Control theory arose from a need to control synthetic sys- uitously exploit regulatory mechanisms for maintaining, con- tems. From regulating steam engines to tuning radios to de- trolling, and adjusting parameters across all scales, from sin- vices capable of autonomous movement, it provided a formal gle molecules to populations of organisms, from microseconds mathematical basis for understanding the role of feedback in to years. These regulatory networks can form functional con- the stability (or change) of dynamical systems. It provides a nections within and across multiple levels (Egiazaryan and framework for understanding any system with feedback regu- Sudakov, 2007). This feedback often radically alters the dy- lation, including biological ones such as regulatory gene net- namic character of the subsystems that comprise a closed-loop works, cellular metabolic systems, sensorimotor dynamics of system, rendering unstable systems stable, fragile systems ro- moving animals, and even ecological or evolutionary dynam- bust, or slow systems fast. Consequently, the properties of ics of organisms and populations. Here we focus on four case individual components (e.g. -
U Ottawa L'universite Canadienne Canada's University T
u Ottawa L'Universite canadienne Canada's university t FACULTE DES ETUDES SUPERIEURES FACULTY OF GRADUATE AND ET POSTOCTORALES u Ottawa POSDOCTORAL STUDIES L'University canadienne Canada's university Mayron Moorhead AUTEUR DE LA THESE / AUTHOR OF THESIS M.Sc. (Biology) GRADE/DEGREE Department of Biology FACULTE, ECOLE, DEPARTEMENT/FACULTY, SCHOOL, DEPARTMENT TITRE DE LA THESE / TITLE OF THESIS Kathleen Gilmour DIRECTEUR (DIRECTRICE) DE LA THESE / THESIS SUPERVISOR John Lewis CO-DIRECTEUR (CO-DIRECTRICE) DE LA THESE/THESIS CO-SUPERVISOR Steve Perry Charles-Antoine Darveau Jayne Yack Gary W. Slater Le Doyen de la Faculte des etudes superieures et postdoctorales / Dean of the Faculty of Graduate and Postdoctoral Studies The Metabolic Cost of Electric Signalling in Weakly Electric Fish By Mayron Moorhead Thesis submitted to the School of Graduate Studies and Research University of Ottawa In partial fulfillment of the requirements for the M.Sc. Degree in the Ottawa-Carleton Institute of biology These soumise a I'Ecole d'etudes superieure et de recherche Universite d'Ottawa envers la realization partielle des exigeances du degree M.Sc. de I'lnstitut de Biologie Ottawa-Carleton Library and Archives Bibliotheque et 1*1 Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington OttawaONK1A0N4 Ottawa ON K1A 0N4 Canada Canada Your file Votre r6terence ISBN: 978-0-494-79672-6 Our file Notre rSfSrence ISBN: 978-0-494-79672-6 NOTICE: AVIS: The author has granted a non L'auteur a accorde