Bacillus Anthracis Edema Factor Substrate Specificity: Evidence for New Modes of Action
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DNA DEAMINATION REPAIR ENZYMES in BACTERIAL and HUMAN SYSTEMS Rongjuan Mi Clemson University, [email protected]
Clemson University TigerPrints All Dissertations Dissertations 12-2008 DNA DEAMINATION REPAIR ENZYMES IN BACTERIAL AND HUMAN SYSTEMS Rongjuan Mi Clemson University, [email protected] Follow this and additional works at: https://tigerprints.clemson.edu/all_dissertations Part of the Biochemistry Commons Recommended Citation Mi, Rongjuan, "DNA DEAMINATION REPAIR ENZYMES IN BACTERIAL AND HUMAN SYSTEMS" (2008). All Dissertations. 315. https://tigerprints.clemson.edu/all_dissertations/315 This Dissertation is brought to you for free and open access by the Dissertations at TigerPrints. It has been accepted for inclusion in All Dissertations by an authorized administrator of TigerPrints. For more information, please contact [email protected]. DNA DEAMINATION REPAIR ENZYMES IN BACTERIAL AND HUMAN SYSTEMS A Dissertation Presented to the Graduate School of Clemson University In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Biochemistry by Rongjuan Mi December 2008 Accepted by: Dr. Weiguo Cao, Committee Chair Dr. Chin-Fu Chen Dr. James C. Morris Dr. Gary Powell ABSTRACT DNA repair enzymes and pathways are diverse and critical for living cells to maintain correct genetic information. Single-strand-selective monofunctional uracil DNA glycosylase (SMUG1) belongs to Family 3 of the uracil DNA glycosylase superfamily. We report that a bacterial SMUG1 ortholog in Geobacter metallireducens (Gme) and the human SMUG1 enzyme are not only uracil DNA glycosylases (UDG), but also xanthine DNA glycosylases (XDG). Mutations at M57 (M57L) and H210 (H210G, H210M, H210N) can cause substantial reductions in XDG and UDG activities. Increased selectivity is achieved in the A214R mutant of Gme SMUG1 and G60Y completely abolishes XDG and UDG activity. Most interestingly, a proline substitution at the G63 position switches the Gme SMUG1 enzyme to an exclusive uracil DNA glycosylase. -

Batrachotoxin Time-Resolved Absorption And
Dental, Oral and Maxillofacial Research Mini Review ISSN: 2633-4291 Batrachotoxin time-resolved absorption and resonance FT-IR and raman biospectroscopy and density functional theory (DFT) investigation of vibronic-mode coupling structure in vibrational spectra analysis: A spectroscopic study on an anti-gum cancer drug Alireza Heidari1,2*, Jennifer Esposito1 and Angela Caissutti1 1Faculty of Chemistry, California South University, 14731 Comet St. Irvine, CA 92604, USA 2American International Standards Institute, Irvine, CA 3800, USA Abstract Gum cancers are cancers that arise from the gum. They are due to the development of abnormal cells that have the ability to invade or spread to other parts of the body. There are three main types of gum cancers: basal-cell gum cancer (BCC), squamous-cell gum cancer (SCC) and melanoma. Batrachotoxin (BTX) is an anti- gum cancer extremely potent cardiotoxic and neurotoxic steroidal alkaloid found in certain species of beetles, birds, and frogs. Batrachotoxin was derived from the Greek word βάτραχος bátrachos "frog". Structurally-related chemical compounds are often referred to collectively as batrachotoxins. It is an extremely poisonous alkaloid. In certain frogs this alkaloid is present mostly on the gum. Such frogs are among those used for poisoning darts. Batrachotoxin binds to and irreversibly opens the sodium channels of nerve cells and prevents them from closing, resulting in paralysis-no antidote is known. Parameters such as FT -IR and Raman vibrational wavelengths and intensities for single crystal Batrachotoxin are calculated using density functional theory and were compared with empirical results. The investigation about vibrational spectrum of cycle dimers in crystal with carboxyl groups from each molecule of acid was shown that it leads to create Hydrogen bonds for adjacent molecules. -
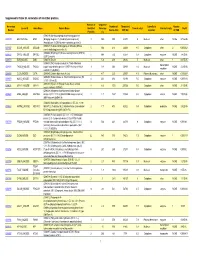
Supporting Table S3 for PDF Maker
Supplemental Table S3. Annotation of identified proteins. Number of Sequence Accession Number of Theoretical Subcellular Number Locus ID Gene Name Protein Name Identified Coverage Theoretical pI Protein Family NSAF Number Amino Acid MW (Da) Location of TMD Peptides (%) (O08539) Myc box-dependent-interacting protein 1 O08539 BIN1_MOUSE BIN1 (Bridging integrator 1) (Amphiphysin-like protein) 3 10.5 588 64470 5 Nucleus other NONE 5.73E-05 (Amphiphysin II) (SH3-domain-containing protein 9) (O08547) Vesicle-trafficking protein SEC22b (SEC22 O08547 SC22B_MOUSE SEC22B 5 30.4 214 24609 8.5 Cytoplasm other 2 0.000262 vesicle-trafficking protein-like 1) (O08553) Dihydropyrimidinase-related protein 2 (DRP-2) O08553 DPYL2_MOUSE DPYSL2 5 19.4 572 62278 6.4 Cytoplasm enzyme NONE 7.85E-05 (ULIP 2 protein) O08579 EMD_MOUSE EMD (O08579) Emerin 1 5.8 259 29436 5 Nucleus other 1 8.67E-05 (O08583) THO complex subunit 4 (Tho4) (RNA and transcription O08583 THOC4_MOUSE THOC4 export factor-binding protein 1) (REF1-I) (Ally of AML-1 1 9.8 254 26809 11.2 Nucleus NONE 2.21E-05 regulator and LEF-1) (Aly/REF) O08585 CLCA_MOUSE CLTA (O08585) Clathrin light chain A (Lca) 2 4.7 235 25557 4.5 Plasma Membrane other NONE 0.000287 (O08600) Endonuclease G, mitochondrial precursor (EC O08600 NUCG_MOUSE ENDOG 4 23.1 294 32191 9.5 Cytoplasm enzyme NONE 0.000134 3.1.30.-) (Endo G) (O08638) Myosin-11 (Myosin heavy chain, smooth O08638 MYH11_MOUSE MYH11 8 6.6 1972 227026 5.5 Cytoplasm other NONE 3.13E-05 muscle isoform) (SMMHC) (O08648) Mitogen-activated protein kinase kinase -

Dependent Protein Kinase to Cyclic CMP Agarose
Binding of Regulatory Subunits of Cyclic AMP- Dependent Protein Kinase to Cyclic CMP Agarose Andreas Hammerschmidt1., Bijon Chatterji2., Johannes Zeiser2, Anke Schro¨ der2, Hans- Gottfried Genieser3, Andreas Pich2, Volkhard Kaever1, Frank Schwede3, Sabine Wolter1", Roland Seifert1*" 1 Institute of Pharmacology, Hannover Medical School, Hannover, Germany, 2 Institute of Toxicology, Hannover Medical School, Hannover, Germany, 3 Biolog Life Science Institute, Bremen, Germany Abstract The bacterial adenylyl cyclase toxins CyaA from Bordetella pertussis and edema factor from Bacillus anthracis as well as soluble guanylyl cyclase a1b1 synthesize the cyclic pyrimidine nucleotide cCMP. These data raise the question to which effector proteins cCMP binds. Recently, we reported that cCMP activates the regulatory subunits RIa and RIIa of cAMP- dependent protein kinase. In this study, we used two cCMP agarose matrices as novel tools in combination with immunoblotting and mass spectrometry to identify cCMP-binding proteins. In agreement with our functional data, RIa and RIIa were identified as cCMP-binding proteins. These data corroborate the notion that cAMP-dependent protein kinase may serve as a cCMP target. Citation: Hammerschmidt A, Chatterji B, Zeiser J, Schro¨der A, Genieser H-G, et al. (2012) Binding of Regulatory Subunits of Cyclic AMP-Dependent Protein Kinase to Cyclic CMP Agarose. PLoS ONE 7(7): e39848. doi:10.1371/journal.pone.0039848 Editor: Andreas Hofmann, Griffith University, Australia Received May 11, 2012; Accepted May 31, 2012; Published July 9, 2012 Copyright: ß 2012 Hammerschmidt et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. -

Mullergliaregnerationtranscriptome
gene.id fc1 fc2 fc3 fc4 p1 p2 p3 p4 FDR.pvalue-1 Gene Symbol Gene Title Pathway go biological process term go molecular function term go cellular component term Dr.10016.1.A1_at -2.33397 -3.86923 -4.38335 -2.39965 0.935201 0.320614 0.208 0.917227 0.208 zgc:77556 zgc:77556 --- proteolysis arylesterase activity /// metallopeptidase activity /// zinc ion --- binding Dr.10024.1.A1 at 1.483417 2.531269 2.089091 1.698761 1 0.613 0.998961 1 0.613 zgc:171808 zgc:171808--- cell-cell signaling --- --- Dr.10051.1.A1 at -1.78449 -2.22024 -1.70922 -1.99464 1 0.901663 1 0.999955 0.9017 ccng2 cyclin G2 --- --- --- --- Dr.10061.1.A1 at 2.065955 2.274632 2.248958 2.507754 0.992 0.718655 0.83 0.600609 0.6006 zgc:173506 zgc:173506--- --- --- --- Dr.10061.2.A1 at 2.131883 2.616483 2.49378 2.815337 0.983443 0.711513 0.805599 0.519115 0.5191 zgc:173506 Zgc:17350 --- --- --- --- Dr.10065.1.A1 at -1.02315 -2.01596 -2.29343 -1.88944 1 0.999955 0.957199 1 0.9572 zgc:114139 zgc:114139--- --- --- cytoplasm /// centrosome Dr.10070.1.A1_at -1.74365 -2.52206 -2.39093 -1.86817 1 0.741254 0.885401 1 0.7413 fbp1a fructose-1, --- carbohydrate metabolic process hydrolase activity /// phosphoric ester hydrolase activity --- Dr.10074.1.S1_at 6.035545 10.44051 7.880519 5.020371 0.1104 0.044 0.062491 0.144679 0.044 pdgfaa platelet-der--- multicellular organismal development /// cell proliferation growth factor activity membrane Dr.10095.1.A1 at -1.73408 -2.11615 -1.47234 -2.19919 1 0.997562 1 0.978177 0.9782 wu:fk95g04 wu:fk95g04--- --- --- --- Dr.10110.1.S1 a at 3.929761 5.798708 -

Triphosphates of Linear-Benzoguanosine, Linear-Benzoinosine, and Linear-Benzoxanthosine
Proc. Natl. Acad. Sci. USA Vol. 76, No. 9, pp. 4262-4264, September 1979 Biochemistry Synthesis of fluorescent nucleotide analogues: 5'-Mono-, di-, and triphosphates of linear-benzoguanosine, linear-benzoinosine, and linear-benzoxanthosine (phosphorylation/xanthine oxidase oxidation/quantum yield/lifetime/dimensional probes) NELSON J. LEONARD AND GENE E. KEYSER Roger Adams Laboratory, School of Chemical Sciences, University of Illinois, Urbana, Illinois 61801 Contributed by Nelson J. Leonard, May 29, 1979 ABSTRACT The fluorescent nucleotide analogues (the 5'- 0 mono-, di-, and triphosphates of lin-benzoguanosine, ftn-ben- 8 9 zoxanthosine, and Iin-benzoinosine) have been prepared for use 7 N 1 as dimensional probes of enzyme binding sites. They have HN N > quantum yields in aqueous solution of 0.39,0.55, and 0.04 and fluorescent lifetimes of 6, 9, and t1.5 nsec, respectively. Un- H2N*J'N 3 Benzoinosine 5'-monophosphate is a substrate for xanthine 5 4 1 oxidase (xanthine:oxygen oxidoreductase, EC 1.2.3.2), providing R lin-benzoxanthosine 5'-monophosphate, and lin-benzoinosine 5'-diphosphate is a substrate for polynucleotide phosphorylase (polyribonucleotide:orthophosphate nucleotidyltransferase, EC 2.7.7.8), giving poly(lin-benzoinosinic acid). The benzologues of the purine diphosphates are substrates for pyruvate kinase (ATP:pyruvate 2-O-phosphotransferase, EC 2.7.1.40), which is I.N used to prepare the triphosphates. Fluorescent analogues or derivatives of adenine-containing O NN 'N nucleotides have been prepared to aid in the definition of en- H I H zyme binding sites and in the determination of interactions in 2 3 nucleic acids (1-6). -

De Novo Nucleic Acids: a Review of Synthetic Alternatives to DNA and RNA That Could Act As † Bio-Information Storage Molecules
life Review De Novo Nucleic Acids: A Review of Synthetic Alternatives to DNA and RNA That Could Act as y Bio-Information Storage Molecules Kevin G Devine 1 and Sohan Jheeta 2,* 1 School of Human Sciences, London Metropolitan University, 166-220 Holloway Rd, London N7 8BD, UK; [email protected] 2 Network of Researchers on the Chemical Evolution of Life (NoR CEL), Leeds LS7 3RB, UK * Correspondence: [email protected] This paper is dedicated to Professor Colin B Reese, Daniell Professor of Chemistry, Kings College London, y on the occasion of his 90th Birthday. Received: 17 November 2020; Accepted: 9 December 2020; Published: 11 December 2020 Abstract: Modern terran life uses several essential biopolymers like nucleic acids, proteins and polysaccharides. The nucleic acids, DNA and RNA are arguably life’s most important, acting as the stores and translators of genetic information contained in their base sequences, which ultimately manifest themselves in the amino acid sequences of proteins. But just what is it about their structures; an aromatic heterocyclic base appended to a (five-atom ring) sugar-phosphate backbone that enables them to carry out these functions with such high fidelity? In the past three decades, leading chemists have created in their laboratories synthetic analogues of nucleic acids which differ from their natural counterparts in three key areas as follows: (a) replacement of the phosphate moiety with an uncharged analogue, (b) replacement of the pentose sugars ribose and deoxyribose with alternative acyclic, pentose and hexose derivatives and, finally, (c) replacement of the two heterocyclic base pairs adenine/thymine and guanine/cytosine with non-standard analogues that obey the Watson–Crick pairing rules. -

Endoq, Exoiii, and Family-4 UDG
www.nature.com/scientificreports OPEN The mesophilic archaeon Methanosarcina acetivorans counteracts uracil in DNA with Received: 21 May 2018 Accepted: 25 September 2018 multiple enzymes: EndoQ, ExoIII, Published: xx xx xxxx and UDG Miyako Shiraishi 1,4,5,6, Sonoko Ishino1, Matthew Hefernan4, Isaac Cann2,3,4,5 & Yoshizumi Ishino1,4,5 Cytosine deamination into uracil is one of the most prevalent and pro-mutagenic forms of damage to DNA. Base excision repair is a well-known process of uracil removal in DNA, which is achieved by uracil DNA glycosylase (UDG) that is found in all three domains of life. However, other strategies for uracil removal seem to have been evolved in Archaea. Exonuclease III (ExoIII) from the euryarchaeon Methanothermobacter thermautotrophicus has been described to exhibit endonuclease activity toward uracil-containing DNA. Another uracil-acting protein, endonuclease Q (EndoQ), was recently identifed from the euryarchaeon Pyrococcus furiosus. Here, we describe the uracil-counteracting system in the mesophilic euryarchaeon Methanosarcina acetivorans through genomic sequence analyses and biochemical characterizations. Three enzymes, UDG, ExoIII, and EndoQ, from M. acetivorans exhibited uracil cleavage activities in DNA with a distinct range of substrate specifcities in vitro, and the transcripts for these three enzymes were detected in the M. acetivorans cells. Thus, this organism appears to conduct uracil repair using at least three distinct pathways. Distribution of the homologs of these uracil-targeting proteins in Archaea showed that this tendency is not restricted to M. acetivorans, but is prevalent and diverse in most Archaea. This work further underscores the importance of uracil- removal systems to maintain genome integrity in Archaea, including ‘UDG lacking’ organisms. -

On the Neurobiology of Physical Activity in Mice and Human in Search of Eating Disorders Determinants
On the Neurobiology of Physical Activity in Mice and Human In Search of Eating Disorders Determinants Elżbieta Kostrzewa ISBN 9789-0393-6090-3 Printed by: CPI Koninklijke Wöhrmann Layout: Elżbieta Kostrzewa and Wouter Wiltenburg Cover design: Elżbieta Kostrzewa and Wouter Wiltenburg More artwork at surrella.wordpress.com . This cover was inspired by The Elephant in the Room , Banksy exhibition, 2006 Barely Legal show, Los Angeles. © Elżbieta Kostrzewa On the Neurobiology of Physical Activity in Mice and Human In Search of Eating Disorders Determinants Over de Neurobiologie van Lichaamsbeweging in Muizen en Mens Op Zoek naar Determinanten van Eetstoornissen (met een samenvatting in het Nederlands) Badania nad neurobiologicznymi podstawami aktywności fizycznej myszy i ludzi W poszukiwaniu determinantów zaburzeń odżywiania (ze streszczeniem w języku polskim) Proefschrift ter verkrijging van de graad van doctor aan de Universiteit Utrecht op gezag van de rector magnificus, prof.dr. G.J. van der Zwaan, ingevolge het besluit van het college voor promoties in het openbaar te verdedigen op donderdag 6 februari 2014 des ochtends te 10.30 uur door Elżbieta Kostrzewa geboren op 10 maart 1983 te Krakau, Polen Promotor: Prof. dr. R.A.H. Adan Co-promotor: Dr. M.J.H. Kas Table of Contents CHAPTER 1 07 General Introduction Physical Activity 08 Eating Disorders 29 Translational Approach in Genetic Studies 43 Scope of the Thesis 49 CHAPTER 2 53 A Candidate Syntenic Genetic Locus is Associated with Physical Activity Levels in Mice and Humans CHAPTER 3 77 -

Anthrax Toxin Time-Resolved Absorption and Resonance Ft-Ir And
Trends in Research Image Article ISSN: 2516-7138 Anthrax toxin time-resolved absorption and resonance ft-ir and raman biospectroscopy and density functional theory (dft) investigation of vibronic-mode coupling structure in vibrational spectra analysis Alireza Heidari1,2*, Jennifer Esposito1 and Angela Caissutti2 1Faculty of Chemistry, California South University, 14731 Comet St. Irvine, CA 92604, USA 2American International Standards Institute, Irvine, CA 3800, USA Abstract Anthrax toxin is a three-protein exotoxin secreted by virulent strains of the bacterium, Bacillus anthracis—the causative agent of anthrax. The toxin was first discovered by Harry Smith in 1954. Anthrax toxin is composed of a cell-binding protein, known as protective antigen (PA), and two enzyme components, called edema factor (EF) and lethal factor (LF). These three protein components act together to impart their physiological effects. Assembled complexes containing the toxin components are endocytosed. In the endosome, the enzymatic components of the toxin translocate into the cytoplasm of a target cell. Once in the cytosol, the enzymatic components of the toxin disrupt various immune cell functions, namely cellular signaling and cell migration. The toxin may even induce cell lysis, as is observed for macrophage cells. Anthrax toxin allows the bacteria to evade the immune system, proliferate, and ultimately kill the host animal. Research on anthrax toxin also provides insight into the generation of macromolecular assemblies, and on protein translocation, pore formation, endocytosis, and other biochemical processes. Parameters such as FT -IR and Raman vibrational wavelengths and intensities for single crystal Anthrax Toxin are calculated using density functional theory and were compared with empirical results. -

Anthrax Toxins in Context of Bacillus Anthracis Spores and Spore Germination
Toxins 2015, 7, 3167-3178; doi:10.3390/toxins7083167 OPEN ACCESS toxins ISSN 2072-6651 www.mdpi.com/journal/toxins Review Anthrax Toxins in Context of Bacillus anthracis Spores and Spore Germination Christopher K. Cote * and Susan L. Welkos United States Army Medical Research Institute of Infectious Diseases (USAMRIID), Bacteriology Division, 1425 Porter Street, Fort Detrick, Frederick, MD 21702-5011, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-301-619-4936; Fax: +1-301-619-2152. Academic Editor: Shihui Liu Received: 29 July 2015 / Accepted: 11 August 2015 / Published: 17 August 2015 Abstract: The interaction of anthrax toxin or toxin components with B. anthracis spores has been demonstrated. Germinating spores can produce significant amounts of toxin components very soon after the initiation of germination. In this review, we will summarize the work performed that has led to our understanding of toxin and spore interactions and discuss the complexities associated with these interactions. Keywords: anthrax; Bacillus anthracis; spores; anthrax toxins; PA; germination 1. Toxins Are Crucial for Bacillus anthracis Pathogenesis Bacillus anthracis, a gram-positive spore-forming bacterium, is the etiologic agent of anthrax [1–4]. It is important to note that, outside of the laboratory, the spore is the only infectious form of B. anthracis [5–8]. In most cases of anthrax acquired by inhalation, the spores are thought to be transported from their site of deposition in the lungs to regional lymph nodes where they germinate and outgrow into the vegetative bacilli [9–15]. -
The Evolving Field of Biodefence: Therapeutic Developments and Diagnostics
REVIEWS THE EVOLVING FIELD OF BIODEFENCE: THERAPEUTIC DEVELOPMENTS AND DIAGNOSTICS James C. Burnett*, Erik A. Henchal‡,Alan L. Schmaljohn‡ and Sina Bavari‡ Abstract | The threat of bioterrorism and the potential use of biological weapons against both military and civilian populations has become a major concern for governments around the world. For example, in 2001 anthrax-tainted letters resulted in several deaths, caused widespread public panic and exerted a heavy economic toll. If such a small-scale act of bioterrorism could have such a huge impact, then the effects of a large-scale attack would be catastrophic. This review covers recent progress in developing therapeutic countermeasures against, and diagnostics for, such agents. BACILLUS ANTHRACIS Microorganisms and toxins with the greatest potential small-molecule inhibitors, and a brief review of anti- The causative agent of anthrax for use as biological weapons have been categorized body development and design against biotoxins is and a Gram-positive, spore- using the scale A–C by the Centers for Disease Control mentioned in TABLE 1. forming bacillus. This aerobic and Prevention (CDC). This review covers the discovery organism is non-motile, catalase and challenges in the development of therapeutic coun- Anthrax toxin. The toxin secreted by BACILLUS ANTHRACIS, positive and forms large, grey–white to white, non- termeasures against select microorganisms and toxins ANTHRAX TOXIN (ATX), possesses the ability to impair haemolytic colonies on sheep from these categories. We also cover existing antibiotic innate and adaptive immune responses1–3,which in blood agar plates. treatments, and early detection and diagnostic strategies turn potentiates the bacterial infection.