Gene Expression Profiling in the Striatum of Per2ko Mice Exhibiting
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Multifactorial Erβ and NOTCH1 Control of Squamous Differentiation and Cancer
Multifactorial ERβ and NOTCH1 control of squamous differentiation and cancer Yang Sui Brooks, … , Karine Lefort, G. Paolo Dotto J Clin Invest. 2014;124(5):2260-2276. https://doi.org/10.1172/JCI72718. Research Article Oncology Downmodulation or loss-of-function mutations of the gene encoding NOTCH1 are associated with dysfunctional squamous cell differentiation and development of squamous cell carcinoma (SCC) in skin and internal organs. While NOTCH1 receptor activation has been well characterized, little is known about how NOTCH1 gene transcription is regulated. Using bioinformatics and functional screening approaches, we identified several regulators of the NOTCH1 gene in keratinocytes, with the transcription factors DLX5 and EGR3 and estrogen receptor β (ERβ) directly controlling its expression in differentiation. DLX5 and ERG3 are required for RNA polymerase II (PolII) recruitment to the NOTCH1 locus, while ERβ controls NOTCH1 transcription through RNA PolII pause release. Expression of several identified NOTCH1 regulators, including ERβ, is frequently compromised in skin, head and neck, and lung SCCs and SCC-derived cell lines. Furthermore, a keratinocyte ERβ–dependent program of gene expression is subverted in SCCs from various body sites, and there are consistent differences in mutation and gene-expression signatures of head and neck and lung SCCs in female versus male patients. Experimentally increased ERβ expression or treatment with ERβ agonists inhibited proliferation of SCC cells and promoted NOTCH1 expression and squamous differentiation both in vitro and in mouse xenotransplants. Our data identify a link between transcriptional control of NOTCH1 expression and the estrogen response in keratinocytes, with implications for differentiation therapy of squamous cancer. Find the latest version: https://jci.me/72718/pdf Research article Multifactorial ERβ and NOTCH1 control of squamous differentiation and cancer Yang Sui Brooks,1,2 Paola Ostano,3 Seung-Hee Jo,1,2 Jun Dai,1,2 Spiro Getsios,4 Piotr Dziunycz,5 Günther F.L. -

Supplementary Materials
Supplementary Materials: Supplemental Table 1 Abbreviations FMDV Foot and Mouth Disease Virus FMD Foot and Mouth Disease NC Non-treated Control DEGs Differentially Expressed Genes RNA-seq High-throughput Sequencing of Mrna RT-qPCR Quantitative Real-time Reverse Transcriptase PCR TCID50 50% Tissue Culture Infective Doses CPE Cytopathic Effect MOI Multiplicity of Infection DMEM Dulbecco's Modified Eagle Medium FBS Fetal Bovine Serum PBS Phosphate Buffer Saline QC Quality Control FPKM Fragments per Kilo bases per Million fragments method GO Gene Ontology KEGG Kyoto Encyclopedia of Genes and Genomes R Pearson Correlation Coefficient NFKBIA NF-kappa-B Inhibitor alpha IL6 Interleukin 6 CCL4 C-C motif Chemokine 4 CXCL2 C-X-C motif Chemokine 2 TNF Tumor Necrosis Factor VEGFA Vascular Endothelial Growth Gactor A CCL20 C-C motif Chemokine 20 CSF2 Macrophage Colony-Stimulating Factor 2 GADD45B Growth Arrest and DNA Damage Inducible 45 beta MYC Myc proto-oncogene protein FOS Proto-oncogene c-Fos MCL1 Induced myeloid leukemia cell differentiation protein Mcl-1 MAP3K14 Mitogen-activated protein kinase kinase kinase 14 IRF1 Interferon regulatory factor 1 CCL5 C-C motif chemokine 5 ZBTB3 Zinc finger and BTB domain containing 3 OTX1 Orthodenticle homeobox 1 TXNIP Thioredoxin-interacting protein ZNF180 Znc Finger Protein 180 ZNF36 Znc Finger Protein 36 ZNF182 Zinc finger protein 182 GINS3 GINS complex subunit 3 KLF15 Kruppel-like factor 15 Supplemental Table 2 Primers for Verification of RNA-seq-detected DEGs with RT-qPCR TNF F: CGACTCAGTGCCGAGATCAA R: -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

A Web-Platform for Analysis of Host Factors Involved in Viral Infections Discovered by Genome Wide Rnai Screen
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2017 vhfRNAi: A web-platform for analysis of host factors involved in viral infections discovered by genome wide RNAi screen Anamika Thakur#, Abid Qureshi# and Manoj Kumar* Bioinformatics Centre, Institute of Microbial Technology, Council of Scientific and Industrial Research, Sector 39A, Chandigarh-160036, India #Equal contribution * To whom correspondence should be addressed. Tel, 91-172-6665453; Fax, 91-172- 2690585; 91-172-2690632; Email, [email protected] Supplementary Tables Table S1: Statistics of unique and duplicate host factors in each virus Table S2: Table denoting genes common among different viruses Table S3: Statistics of GWAS analysis Table S1. Statistics of unique and duplicate host factors in each virus S. No. Virus Unique-Entries Duplicate-Entries 1. Adeno-associated virus (AAV) 926 533 2. Avian influenza virus (AIV) 0 11 3. Borna disease virus (BDV) 14 20 4. Dengue virus 2 (DEN-2) 27 13 5. Hepatitis C virus (HCV) 236 213 6. Human immunodeficiency virus 1 (HIV) 1388 857 7. Human parainfluenza virus 3 (HPIV-3) 0 27 8. Human herpesvirus 1 (HSV-1) 34 38 9. Influenza A virus (IAV) 700 513 10. Lymphocytic choriomeningitis virus (LCMV) 0 54 11. Marburgvirus (MARV) 0 11 12. Poliovirus (PV) 3340 1035 13. Rotavirus (RV) 347 175 14. Sendai virus (SeV) 32 27 15. Sindbis virus (SIV) 70 41 16. Vaccinia virus (VACV) 482 296 17. Vesicular stomatitis virus (VSV) 9 78 18. West Nile virus (WNV) 313 137 Table S1. Statistics of unique host factors for each virus having overlapping and unique factors in different viruses Non-overlap – Overall- S. -

Identification of Hair Shaft Progenitors That Create a Niche for Hair Pigmentation
Downloaded from genesdev.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press Identification of hair shaft progenitors that create a niche for hair pigmentation Chung-Ping Liao,1 Reid C. Booker,1 Sean J. Morrison,2,3,4,5,6 and Lu Q. Le1,4,5 1Department of Dermatology, 2Department of Pediatrics, 3Children’s Research Institute, 4Simmons Comprehensive Cancer Center, 5Hamon Center for Regenerative Science and Medicine, 6Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA Hair differentiates from follicle stem cells through progenitor cells in the matrix. In contrast to stem cells in the bulge, the identities of the progenitors and the mechanisms by which they regulate hair shaft components are poorly understood. Hair is also pigmented by melanocytes in the follicle. However, the niche that regulates follicular melanocytes is not well characterized. Here, we report the identification of hair shaft progenitors in the matrix that are differentiated from follicular epithelial cells expressing transcription factor KROX20. Depletion of Krox20 lin- eage cells results in arrest of hair growth, confirming the critical role of KROX20+ cells as antecedents of structural cells found in hair. Expression of stem cell factor (SCF) by these cells is necessary for the maintenance of differen- tiated melanocytes and for hair pigmentation. Our findings reveal the identities of hair matrix progenitors that regulate hair growth and pigmentation, partly by creating an SCF-dependent niche for follicular melanocytes. [Keywords: stem cell factor (SCF); hair pigmentation; hair shaft progenitor cell; hair follicle stem cell; hair matrix; KROX20] Supplemental material is available for this article. -

Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals
animals Review Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals Mohammadreza Mohammadabadi 1 , Farhad Bordbar 1,* , Just Jensen 2 , Min Du 3 and Wei Guo 4 1 Department of Animal Science, Faculty of Agriculture, Shahid Bahonar University of Kerman, Kerman 77951, Iran; [email protected] 2 Center for Quantitative Genetics and Genomics, Aarhus University, 8210 Aarhus, Denmark; [email protected] 3 Washington Center for Muscle Biology, Department of Animal Sciences, Washington State University, Pullman, WA 99163, USA; [email protected] 4 Muscle Biology and Animal Biologics, Animal and Dairy Science, University of Wisconsin-Madison, Madison, WI 53558, USA; [email protected] * Correspondence: [email protected] Simple Summary: Skeletal muscle mass is an important economic trait, and muscle development and growth is a crucial factor to supply enough meat for human consumption. Thus, understanding (candidate) genes regulating skeletal muscle development is crucial for understanding molecular genetic regulation of muscle growth and can be benefit the meat industry toward the goal of in- creasing meat yields. During the past years, significant progress has been made for understanding these mechanisms, and thus, we decided to write a comprehensive review covering regulators and (candidate) genes crucial for muscle development and growth in farm animals. Detection of these genes and factors increases our understanding of muscle growth and development and is a great help for breeders to satisfy demands for meat production on a global scale. Citation: Mohammadabadi, M.; Abstract: Farm-animal species play crucial roles in satisfying demands for meat on a global scale, Bordbar, F.; Jensen, J.; Du, M.; Guo, W. -

Features of Mirna Binding Sites in Mrnas of Bos Taurus ZNF Family Transcription Factors
ICBMB 2016 International Conference on Biochemistry and Molecular Biology 27-29 April 2016, Venice, Italy Best Western Hotel Bologna Hall Tercicore Via Piave, 214, 30171 Venezia Mestre, Venezia VE, Italy Features of miRNA binding sites in mRNAs of Bos taurus ZNF family transcription factors Aigul Alybayeva1*, Raigul Niyazova1, Bernard Faye2, and Anatoly Ivashchenko1 1al-Farabi KazNU, Almaty, Kazakhstan, *[email protected]. 2CIRAD-ES Campus International de Baillarguet, 34398 Montpellier Cedex 1, France Abstract: miRNA binding sites in the mRNA nucleotide sequences of 247 transcription factor (TF) genes of Bos taurus ZNF family was established. miRNAs binding sites are located in the 5'UTRs, CDSs and 3'UTRs of mRNAs. miRNA binding sites in mRNAs were selected with the value ΔG/ΔGm equal or greater than 85%. miR-574 was identified from 749 Bos taurus miRNAs, it has binding sites with mRNAs of KLF7, SNAI2, WIZ, ZFP1, ZFP91-1, ZNF175, ZNF6772, ZSCAN29-1 genes. The value ΔG/ΔGm varied from 85 to 95%. The starts of miR-574 binding sites with mRNAs of KLF7, ZFP91-1, ZNF6772 genes are located through two nucleotides. miR-574 binding sites are located in the 3'UTR. miR-1260b has binding sites with mRNAs of 29 genes (BCL11B-1, CTCF-1, DPF2-1, EGR3, FEZF2, PRDM13, PRDM14, PRDM6, SALL1, SP6-2, SP8, WIZ, ZFX-1, ZFX-2, ZKSCAN1, ZNF219, ZNF335, ZNF341, ZNF384, ZNF48, ZNF513, ZNF618-1, ZNF653, ZNF668, ZNF746, ZNF75, ZNF805, ZNF774, ZSCAN20-1) which are located in the 3'UTRs, CDSs and 5'UTRs. The value ΔG/ΔGm varied from 85 to 91%. The starts of miR- 1260b binding sites in mRNAs of ZNF618-1 target gene are located through one nucleotide in the 5'UTR and with mRNAs of ZNF384, BCL11B-1 genes through three nucleotides in the CDSs. -

Egr2 and 3 Control Adaptive Immune Responses by Temporally Uncoupling Expansion from T Cell Differentiation
Published Online: 9 May, 2017 | Supp Info: http://doi.org/10.1084/jem.20160553 Downloaded from jem.rupress.org on October 9, 2018 Article Egr2 and 3 control adaptive immune responses by temporally uncoupling expansion from T cell differentiation Tizong Miao,1* Alistair L.J. Symonds,1* Randeep Singh,1,2 Janine D. Symonds,3 Ane Ogbe,2 Becky Omodho,2 Bo Zhu,1,4 Suling Li,2 and Ping Wang1 1The Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, England, UK 2Bioscience, Brunel University, Uxbridge UB8 3PH, England, UK 3Centre for Mathematics and Physics in the Life Sciences and Experimental Biology (CoMPLEX), University College London, London WC1E 6BT, England, UK 4Institute of Cancer, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, People’s Republic of China Egr2 and 3 are important for maintaining immune homeostasis. Here we define a fundamental function of Egr2 and 3 oper- ating as a checkpoint that controls the transition between clonal expansion and differentiation of effector T cells. Egr2 and 3 deficiency resulted in defective clonal expansion but hyperactivation and excessive differentiation of T cells in response to viral infection. Conversely, sustained Egr2 expression enhanced expansion but severely impaired effector differentiation. Egr2 bound to and controlled the expression of genes regulating proliferation (Myc and Myb) and differentiation repressors (Bcl6, Id3), while repressing transcription factors required for effector function (Zeb2, RORa, RORc, and Bhlhe40). Egr2 and 3 ex- pression in T cells was regulated reciprocally by antigen and IFNγ, providing a mechanism for adjusting proliferation and dif- ferentiation of individual T cells. -
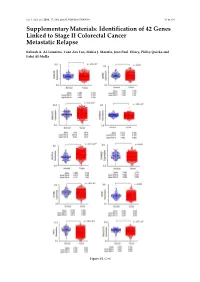
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN -

Loss of the Neural-Specific BAF Subunit ACTL6B Relieves Repression of Early Response Genes and Causes Recessive Autism
Loss of the neural-specific BAF subunit ACTL6B relieves repression of early response genes and causes recessive autism Wendy Wenderskia,b,c,d, Lu Wange,f,g,1, Andrey Krokhotina,b,c,d,1, Jessica J. Walshh, Hongjie Lid,i, Hirotaka Shojij, Shereen Ghoshe,f,g, Renee D. Georgee,f,g, Erik L. Millera,b,c,d, Laura Eliasa,b,c,d, Mark A. Gillespiek, Esther Y. Sona,b,c,d, Brett T. Staahla,b,c,d, Seung Tae Baeke,f,g, Valentina Stanleye,f,g, Cynthia Moncadaa,b,c,d, Zohar Shiponya,b,c,d, Sara B. Linkerl, Maria C. N. Marchettol, Fred H. Gagel, Dillon Chene,f,g, Tipu Sultanm, Maha S. Zakin, Jeffrey A. Ranishk, Tsuyoshi Miyakawaj, Liqun Luod,i, Robert C. Malenkah, Gerald R. Crabtreea,b,c,d,2, and Joseph G. Gleesone,f,g,2 aDepartment of Pathology, Stanford Medical School, Palo Alto, CA 94305; bDepartment of Genetics, Stanford Medical School, Palo Alto, CA 94305; cDepartment of Developmental Biology, Stanford Medical School, Palo Alto, CA 94305; dHoward Hughes Medical Institute, Stanford University, Palo Alto, CA 94305; eDepartment of Neuroscience, University of California San Diego, La Jolla, CA 92037; fHoward Hughes Medical Institute, University of California San Diego, La Jolla, CA 92037; gRady Children’s Institute of Genomic Medicine, University of California San Diego, La Jolla, CA 92037; hNancy Pritztker Laboratory, Department of Psychiatry and Behavioral Sciences, Stanford Medical School, Palo Alto, CA 94305; iDepartment of Biology, Stanford University, Palo Alto, CA 94305; jDivision of Systems Medical Science, Institute for Comprehensive Medical Science, Fujita Health University, 470-1192 Toyoake, Aichi, Japan; kInstitute for Systems Biology, Seattle, WA 98109; lLaboratory of Genetics, The Salk Institute for Biological Studies, La Jolla, CA 92037; mDepartment of Pediatric Neurology, Institute of Child Health, Children Hospital Lahore, 54000 Lahore, Pakistan; and nClinical Genetics Department, Human Genetics and Genome Research Division, National Research Centre, 12311 Cairo, Egypt Edited by Arthur L. -

Egr2/Egr3 Are Essential Tumour Suppressor Genes for Lymphomagenesis
Egr2/Egr3 are Essential Tumour Suppressor Genes for Lymphomagenesis A thesis submitted for the degree of Doctor of Philosophy by Punamdip Kaur Bhullar September 2013 Division of Biosciences School of Health Sciences and Social Care Declaration I hereby declare that the research presented in this thesis is my own work, except where otherwise specified, and has not been submitted for any other degree. Punamdip Kaur Bhullar II Abstract Non-Hodgkin’s lymphoma is the fifth most common cancer in the UK, accounting for 4% of all new cases. The control of lymphomagenesis still remains a challenge. Early growth response gene (Egr) 2 and 3 are zinc finger transcription factors. Egr2 plays an important role in the development of both central nervous system and lymphocytes. However the mechanism of action in lymphocytes is still unknown. In order to fully understand the function of Egr2, in lymphocytes, we developed Egr2 and 3 double knockout mice (Egr2-/-Egr3-/-) by crossbreeding lymphocyte specific Egr2 knockout mice (CD2-Egr2-/-) with Egr3 knockout mice (Egr3-/-), as previous reports suggested that Egr3 compensates for the role of Egr2. In the absence of Egr2 and 3, the homeostasis of T cells is dysregulated with hyper- homeostatic proliferation of effector like phenotype cells. More importantly the development of spontaneous B and T cell lymphoma was found in more than 70% of Egr2-/-Egr3-/- mice. The lymphoma cells from Egr2-/-Egr3-/- mice were highly proliferative and metastatically spread into other non-lymphoid organs, such as lung, liver and kidney. In additional to this lymphoma development the Egr2-/-Egr3-/- mice showed signs of chronic inflammatory disorder. -

Analysis of Light-Induced Immediate-Early Gene Expression in the Suprachiasmatic Nucleus
ANALYSIS OF LIGHT-INDUCED IMMEDIATE-EARLY GENE EXPRESSION IN THE SUPRACHIASMATIC NUCLEUS A thesis submitted to Kent State University in partial fulfillment of the requirements for the degree of Master of Science By, Amanda S. Ohnmeiss August 2009 Thesis written by Amanda S. Ohnmeiss B.S. Allegheny College, 2007 M.S., Kent State University, 2009 Approved by _____ Dr. E. Mintz _______________, Advisor ____ Dr. R. Dorman ______________, Director, School of Biomedical Sciences ___Dr. T. Moerland______________, Dean, College of Arts and Sciences ii TABLE OF CONTENTS PAGE LIST OF FIGURES ………………………………………………………………………v LIST OF TABLES………………………………………………………….………….…vi ACKNOWLEDGEMENTS………………………………………………………..…….vii Introduction……………………………………………………….........................1 Suprachiasmatic Nucleus Background…………………........................................1 Entrainment………………………………………………………………………..2 Common Light-Induced Genes of the SCN……………………………………….3 Glutamate………………………………………...………………………..8 Goals………………………………………………………………………9 Methods…………………………………………………………………….…….13 Animals.....................................................................................................13 Experiment 1……………………………………………………………..13 Experiment 2……………………………………………………………..14 Experiment 3………………………………………………………….….14 Experiment 4………………………………………………………….….15 Immunohistochemistry……………………………………………..……15 Data Analysis…………………………………………………………….16 Results…………………………………………………………………………....18 iii Discussion………………………………………………………………………..38 References……………………………………………………………………….47 iv LIST OF FIGURES