Sequestration of a Highly Reactive Intermediate in an Evolving Pathway for Degradation of Pentachlorophenol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ferric Reductase Activity of the Arsh Protein from Acidithiobacillus Ferrooxidans
J. Microbiol. Biotechnol. (2011), 21(5), 464–469 doi: 10.4014/jmb.1101.01020 First published online 13 April 2011 Ferric Reductase Activity of the ArsH Protein from Acidithiobacillus ferrooxidans Mo, Hongyu1,2, Qian Chen1,2, Juan Du1, Lin Tang1, Fang Qin1, Bo Miao2, Xueling Wu2, and Jia Zeng1,2* 1College of Biology, Hunan University, Changsha, Hunan 410082, P. R. China 2Department of Bioengineering, Central South University, Changsha, Hunan 410083, P. R. China Received: January 14, 2011 / Revised: March 10, 2011 / Accepted: March 11, 2011 The arsH gene is one of the arsenic resistance system in iron (free or chelated) into ferrous iron before its incorporation bacteria and eukaryotes. The ArsH protein was annotated into heme and nonheme iron-containing proteins. Ferric as a NADPH-dependent flavin mononucleotide (FMN) reductase catalyzes the reduction of complexed Fe3+ to reductase with unknown biological function. Here we complexed Fe2+ using NAD(P)H as the electron donor. The report for the first time that the ArsH protein showed resulting Fe2+ is subsequently released and incorporated high ferric reductase activity. Glu104 was an essential into iron-containing proteins [17]. residue for maintaining the stability of the FMN cofactor. Here we report for the first time that the ArsH protein The ArsH protein may perform an important role for showed high ferric reduction activity. The ArsH from A. cytosolic ferric iron assimilation in vivo. ferrooxidans may perform an important role as a NADPH- Keywords: Acidithiobacillus ferrooxidans, ArsH, flavoprotein, dependent ferric reductase for cytosolic ferric iron assimilation ferric reductase in vivo. MATERIALS AND METHODS Arsenic resistance genes are widespread in nature. -
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Anamika Kothari Quang Ong Ron Caspi An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Peter D Karp Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Csac1394711Cyc: Candidatus Saccharibacteria bacterium RAAC3_TM7_1 Cellular Overview Connections between pathways are omitted for legibility. Tim Holland TM7C00001G0420 TM7C00001G0109 TM7C00001G0953 TM7C00001G0666 TM7C00001G0203 TM7C00001G0886 TM7C00001G0113 TM7C00001G0247 TM7C00001G0735 TM7C00001G0001 TM7C00001G0509 TM7C00001G0264 TM7C00001G0176 TM7C00001G0342 TM7C00001G0055 TM7C00001G0120 TM7C00001G0642 TM7C00001G0837 TM7C00001G0101 TM7C00001G0559 TM7C00001G0810 TM7C00001G0656 TM7C00001G0180 TM7C00001G0742 TM7C00001G0128 TM7C00001G0831 TM7C00001G0517 TM7C00001G0238 TM7C00001G0079 TM7C00001G0111 TM7C00001G0961 TM7C00001G0743 TM7C00001G0893 TM7C00001G0630 TM7C00001G0360 TM7C00001G0616 TM7C00001G0162 TM7C00001G0006 TM7C00001G0365 TM7C00001G0596 TM7C00001G0141 TM7C00001G0689 TM7C00001G0273 TM7C00001G0126 TM7C00001G0717 TM7C00001G0110 TM7C00001G0278 TM7C00001G0734 TM7C00001G0444 TM7C00001G0019 TM7C00001G0381 TM7C00001G0874 TM7C00001G0318 TM7C00001G0451 TM7C00001G0306 TM7C00001G0928 TM7C00001G0622 TM7C00001G0150 TM7C00001G0439 TM7C00001G0233 TM7C00001G0462 TM7C00001G0421 TM7C00001G0220 TM7C00001G0276 TM7C00001G0054 TM7C00001G0419 TM7C00001G0252 TM7C00001G0592 TM7C00001G0628 TM7C00001G0200 TM7C00001G0709 TM7C00001G0025 TM7C00001G0846 TM7C00001G0163 TM7C00001G0142 TM7C00001G0895 TM7C00001G0930 Detoxification Carbohydrate Biosynthesis DNA combined with a 2'- di-trans,octa-cis a 2'- Amino Acid Degradation an L-methionyl- TM7C00001G0190 superpathway of pyrimidine deoxyribonucleotides de novo biosynthesis (E. -

The Human Flavoproteome
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Archives of Biochemistry and Biophysics 535 (2013) 150–162 Contents lists available at SciVerse ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi Review The human flavoproteome ⇑ Wolf-Dieter Lienhart, Venugopal Gudipati, Peter Macheroux Graz University of Technology, Institute of Biochemistry, Petersgasse 12, A-8010 Graz, Austria article info abstract Article history: Vitamin B2 (riboflavin) is an essential dietary compound used for the enzymatic biosynthesis of FMN and Received 17 December 2012 FAD. The human genome contains 90 genes encoding for flavin-dependent proteins, six for riboflavin and in revised form 21 February 2013 uptake and transformation into the active coenzymes FMN and FAD as well as two for the reduction to Available online 15 March 2013 the dihydroflavin form. Flavoproteins utilize either FMN (16%) or FAD (84%) while five human flavoen- zymes have a requirement for both FMN and FAD. The majority of flavin-dependent enzymes catalyze Keywords: oxidation–reduction processes in primary metabolic pathways such as the citric acid cycle, b-oxidation Coenzyme A and degradation of amino acids. Ten flavoproteins occur as isozymes and assume special functions in Coenzyme Q the human organism. Two thirds of flavin-dependent proteins are associated with disorders caused by Folate Heme allelic variants affecting protein function. Flavin-dependent proteins also play an important role in the Pyridoxal 50-phosphate biosynthesis of other essential cofactors and hormones such as coenzyme A, coenzyme Q, heme, pyri- Steroids doxal 50-phosphate, steroids and thyroxine. Moreover, they are important for the regulation of folate Thyroxine metabolites by using tetrahydrofolate as cosubstrate in choline degradation, reduction of N-5.10-meth- Vitamins ylenetetrahydrofolate to N-5-methyltetrahydrofolate and maintenance of the catalytically competent form of methionine synthase. -

Reaction Mechanism of Azoreductases Suggests Convergent Evolution with Quinone Oxidoreductases
Protein Cell 2010, 1(8): 780–790 Protein & Cell DOI 10.1007/s13238-010-0090-2 RESEARCH ARTICLE Reaction mechanism of azoreductases suggests convergent evolution with quinone oxidoreductases ✉ Ali Ryan*, Chan-Ju Wang*, Nicola Laurieri*, Isaac Westwood, Edith Sim Department of Pharmacology, University of Oxford, Mansfield Road, Oxford, OX1 3QT, UK ✉ Correspondence: [email protected] Received June 2, 2010 Accepted June 28, 2010 ABSTRACT the dye used for dying fabrics does not bind to the fabric, and as a result, is lost in the effluent (Shore, 1995). As these dyes Azoreductases are involved in the bioremediation by can be carcinogenic (Alves de Lima et al., 2007) their removal bacteria of azo dyes found in waste water. In the gut flora, from the effluent is essential. This has driven investigations they activate azo pro-drugs, which are used for treatment into both electrochemical reduction of azo dyes (Wang et al., of inflammatory bowel disease, releasing the active 2010b), as well as the use of both aerobic and anaerobic component 5-aminosalycilic acid. The bacterium bacteria for bioremediation (dos Santos et al., 2007; You et P. aeruginosa has three azoreductase genes, paAzoR1, al., 2007). paAzoR2 and paAzoR3, which as recombinant enzymes Azo bonds are also found in some drugs including those have been shown to have different substrate specifici- used for the treatment of inflammatory bowel disease (IBD). ties. The mechanism of azoreduction relies upon tauto- Azo compounds have also been shown to be effective merisation of the substrate to the hydrazone form. We antibacterial (Farghaly and Abdalla, 2009) and antitumor (El- report here the characterization of the P. -
Q 297 Suppl USE
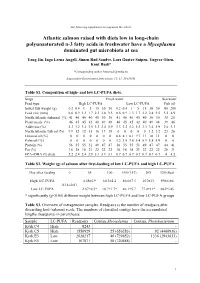
The following supplement accompanies the article Atlantic salmon raised with diets low in long-chain polyunsaturated n-3 fatty acids in freshwater have a Mycoplasma dominated gut microbiota at sea Yang Jin, Inga Leena Angell, Simen Rød Sandve, Lars Gustav Snipen, Yngvar Olsen, Knut Rudi* *Corresponding author: [email protected] Aquaculture Environment Interactions 11: 31–39 (2019) Table S1. Composition of high- and low LC-PUFA diets. Stage Fresh water Sea water Feed type High LC-PUFA Low LC-PUFA Fish oil Initial fish weight (g) 0.2 0.4 1 5 15 30 50 0.2 0.4 1 5 15 30 50 80 200 Feed size (mm) 0.6 0.9 1.3 1.7 2.2 2.8 3.5 0.6 0.9 1.3 1.7 2.2 2.8 3.5 3.5 4.9 North Atlantic fishmeal (%) 41 40 40 40 40 30 30 41 40 40 40 40 30 30 35 25 Plant meals (%) 46 45 45 42 40 49 48 46 45 45 42 40 49 48 39 46 Additives (%) 3.3 3.2 3.2 3.5 3.3 3.4 3.9 3.3 3.2 3.2 3.5 3.3 3.4 3.9 2.6 3.3 North Atlantic fish oil (%) 9.9 12 12 15 16 17 18 0 0 0 0 0 1.2 1.2 23 26 Linseed oil (%) 0 0 0 0 0 0 0 6.8 8.1 8.1 9.7 11 10 11 0 0 Palm oil (%) 0 0 0 0 0 0 0 3.2 3.8 3.8 5.4 5.9 5.8 5.9 0 0 Protein (%) 56 55 55 51 49 47 47 56 55 55 51 49 47 47 44 41 Fat (%) 16 18 18 21 22 22 22 16 18 18 21 22 22 22 28 31 EPA+DHA (% diet) 2.2 2.4 2.4 2.9 3.1 3.1 3.1 0.7 0.7 0.7 0.7 0.7 0.7 0.7 4 4.2 Table S2. -

Probing the Flavin Transfer Mechanism in Alkanesulfonate Monooxygenase System
bioRxiv preprint doi: https://doi.org/10.1101/433839; this version posted October 2, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Probing the flavin transfer mechanism in alkanesulfonate monooxygenase system Dayal PV and Ellis HR Department of Chemistry and Biochemistry, Auburn University, Alabama, 36849 Email: [email protected] Abstract Bacteria acquire sulfur through the sulfur assimilation pathway, but under sulfur limiting conditions bacteria must acquire sulfur from alternative sources. The alkanesulfonate monooxygenase enzymes are expressed under sulfur-limiting conditions, and catalyze the desulfonation of wide-range of alkanesulfonate substrates. The SsuE enzyme is an NADPH-dependent FMN reductase that provides reduced flavin to the SsuD monooxygenase. The mechanism for the transfer of reduced flavin in flavin dependent two-component systems occurs either by free- diffusion or channeling. Previous studies have shown the presence of protein-protein interactions between SsuE and SsuD, but the identification of putative interaction sights have not been investigated. Current studies utilized HDX-MS to identify protective sites on SsuE and SsuD. A conserved α-helix on SsuD showed a decrease in percent deuteration when SsuE was included in the reaction. This suggests the role of α-helix in promoting protein-protein interactions. Specific SsuD variants were generated in order to investigate the role of these residues in protein-protein interactions and catalysis. Variant containing substitutions at the charged residues showed a six-fold decrease in the activity, while a deletion variant of SsuD lacking the α-helix showed no activity when compared to wild-type SsuD. -

Structural Features That Promote Catalysis in Two-Component Systems Involved in Sulfur Metabolism
Structural Features That Promote Catalysis in Two-Component Systems Involved in Sulfur Metabolism by Richard Allen Hagen A dissertation to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 8, 2020 Copyright 2020 by Richard Allen Hagen Approved by Holly R. Ellis, Chair, Professor of Chemistry and Biochemistry Douglas C. Goodwin, Professor of Chemistry and Biochemistry Evert C. Duin, Professor of Chemistry and Biochemistry Steven Mansoorabadi, Associate Professor of Chemistry and Biochemistry Abstract Sulfur is an essential element important in the synthesis of biomolecules. Bacteria are able to assimilate inorganic sulfur for the biosynthesis of L-cysteine. Inorganic sulfate is often unavailable, so bacteria have evolved multiple metabolic pathways to obtain sulfur from alternative sources. Interestingly, many of the enzymes involved in sulfur acquisition are flavin- dependent two-component systems. These two-component systems consist of a flavin reductase and monooxygenase that utilize flavin to cleave the carbon-sulfur bonds of organosulfur compounds. The two-component systems differ in their characterized sulfur substrate specificity. Enzymes SsuE/SsuD are involved in the desulfonation of linear alkanesulfonates (C2-C10), enzymes MsuE/MsuD utilize methanesulfonate (C1), and enzymes SfnF/SfnG utilize DMSO2 as a sulfur source. The flavin reductases involved in sulfur assimilation utilize FMN as a substrate but differ in their ability to utilize NADH or NADPH. The alkanesulfonate monooxygenase system was the first two-component flavin-dependent system expressed during sulfur limiting conditions that was characterized. The flavin reductase (SsuE) and monooxygenase (SsuD) have distinct structural and functional properties, but the two enzymes must synchronize their functions for catalysis to occur. -

Novocib E-Nov8
PRECICE ® Services Information sheet Ref: # E-Nov 8 Bacterial FMN-reductase from Escherichia coli (Fre) Synonyms: NAD(P)H:flavin oxidoreductase, NAD(P)H:flavin mono- nucleotide oxidoreductase, NAD(P)H(2):FMN oxidoreductase, NAD(P)H-FMN reductase, NAD(P)H-dependent FMN reductase, NAD(P)H:FMN oxidoreductase, riboflavin mononucleotide reductase, flavin mononucleotide reductase E.C. 1.5.1.29 Description NOVO CIB 's bacterial (E. coli ) NAD(P)H-dependent FMN-oxidoreductase is a recombinant protein of ca.26kDa overexpressed in E.coli . The sequence of cloned Fre (SwissProt accession number P0AEN1) was confirmed by DNA sequencing (100% identity). NAD(P)H:flavin oxidoreductases (or flavin reductases) catalyze the reduction of riboflavin, FMN, and FAD by NADPH and NADH, and are present in all microorganisms, including the luminous marine bacteria and also in mammals. Flavin reductases have been classified into two groups. The first group includes flavoproteins that use a flavin prosthetic group for the electron transfer from NAD(P)H to the flavin substrate (flavin reductase P from Vibrio harveyi ). In the second group, enzymes do not contain any prosthetic group. Fre, NAD(P)H:flavin oxidoreductase from E. coli , belongs to this second group of enzymes. Applications Coupling of bacterial luciferase to FMN-NAD(P)H oxidoreductase has been used to provide ultrasensitive analytical tools for the quantification of NADH and the substrates of NADH-, NADPH- dependent enzymes ( e.g. glucose, lactate, malate, ethanol, sorbitol, oxaloacetate). Although FMN-reductase often present in luciferase enzyme preparations may be sufficient for producing light in the presence of NAD(P)H, highly purified and characterized Fre enzyme can offer some advantages such as an increased sensitivity, better control of the signal intensity and duration, and saving of the luciferase enzyme (Figure 1). -

Subject Index to Volume 73 JANUARY-DE(MEMBER 1976
Subject Index to Volume 73 JANUARY-DE(MEMBER 1976 Introduction The terms in the Subject Index for Vol ume 73, January-December 1976, of the PROCEEDINGSOF THE NATIONALACA DEMY OF SCIENCESUSA were chosen mainly from titles and key terms of artic] es. The index terms are alphabetized by computer; numbers, conformational pref ixes, Greek letters, hyphens, and spaces between words are disregarded in alphabe tization. After each index term is printed the title of the article (or a suitable modi fication of the title) and the appropriate page number. Titles are listed in alphabe tical order under the index terms. Classifications (e.g., Physics, Mather latics) under which papers have been published are used as index terms only if it seemed this would be helpful. Papers that are concerned in some major way with methodology are indexed under "Methodology" as well as under more sj ,ecific terms. Papers relevant to human diseases are indexed under "Diseases of I iuman beings" (with some subclassifica- tions) as well as under more specific tern is. Corrections to papers in which errors occurred are indexed under the term "Coi rection" as well as under the index terms selected for the paper itself. Organisms are indexed by their scientific names when scientific names were provided in the pape rs; suitable cross-references are provided. Genes are listed together as, for example ,"Gene, lac." Because the PROCEEDINGSurges auth ors to follow the tentative rules and rec- ommendations of the nomenclature cor amissions (e.g., for biochemistry, those proposed by the International Union ol 'Pure and Applied Chemistry and the Commission on Biochemical Nomenclatu re), an effort has been made to construct an index that conforms with this policy. -

Tenebrionid Secretions and a Fungal Benzoquinone Oxidoreductase Form
Tenebrionid secretions and a fungal benzoquinone PNAS PLUS oxidoreductase form competing components of an arms race between a host and pathogen Nicolás Pedrinia,1,2, Almudena Ortiz-Urquizab,1, Carla Huarte-Bonneta, Yanhua Fanb,c, M. Patricia Juáreza, and Nemat O. Keyhanib,2 SEE COMMENTARY aInstituto de Investigaciones Bioquímicas de La Plata (Centra Cientifico Technologia La Plata Consejo Nacional de Investigaciones Científicas y Técnicas-Universidad Nacional de La Plata), Facultad de Ciencias Médicas, La Plata 1900, Argentina; bDepartment of Microbiology and Cell Science, Institute of Food and Agricultural Sciences, University of Florida, Gainesville, FL 32611; and cBiotechnology Research Center, Southwest University, Beibei, Chongqing, 400715, China Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved May 19, 2015 (received for review March 9, 2015) Entomopathogenic fungi and their insect hosts represent a model and maintenance of sex, genetic recombination, and immune sys- system for examining invertebrate-pathogen coevolutionary se- tems (3–5); (iii) as a framework for understanding invasions of lection processes. Here we report the characterization of compet- exotic species (6); and (iv)asmechanism(s)selectingforhost– ing components of an arms race consisting of insect protective pathogen coevolution [i.e., broad versus specific host ranges and/or antimicrobial compounds and evolving fungal mechanisms of de- host tolerance (7–9)]. Beauveria bassiana toxification. The insect pathogenic fungus has For fungal pathogens, the best-known examples of coevolu- a remarkably wide host range; however, some insects are resistant tionary interactions are the pathogen effector and corresponding to fungal infection. Among resistant insects is the tenebrionid plant host target genes that have defined a gene-for-gene disease Tribolium castaneum beetle that produces benzoquinone-contain- susceptibility model in which the outcome of infection is based ing defensive secretions. -

Exploring the Functional Roles of Π-Helices and Flavin-Induced Oligomeric State Changes in Flavin Reductases of Two-Component Monooxygenase Systems
Exploring the Functional Roles of π-helices and Flavin-Induced Oligomeric State Changes in Flavin Reductases of Two-Component Monooxygenase Systems by Jonathan Makau Musila A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama May 6, 2017 Keywords: Oligomerization, kinetics, desulfonation, π-helix, dis(asso)ciation Copyright 2017 by Jonathan Makau Musila Approved by Holly R. Ellis, Chair, Professor of Chemistry and Biochemistry Douglas C. Goodwin, Associate Professor of Chemistry and Biochemistry Evert C. Duin, Associate Professor of Chemistry and Biochemistry Steven Mansoorabadi, Assistant Professor of Chemistry and Biochemistry Abstract Sulfur-containing biomolecules participate in various chemical and structural functions in enzymes. When sulfate is limiting, bacteria upregulate ssi enzymes to utilize organosulfonates as an alternative source. The alkanesulfonate monooxygenase enzymes mitigate sulfur scarcity through desulfonation of various alkanesulfonates releasing sulfite which is incorporated into sulfur-containing biomolecules. This two-component monooxygenase system utilizes flavin as a substrate with the SsuE enzyme supplying reduced flavin to SsuD. It is unclear what structural properties of flavin reductases of two-component systems dictate catalysis. The SsuE enzyme undergoes a tetramer to dimer oligomeric switch in the presence of FMN. Oligomeric state changes are common in flavin reductases but the roles and regulation of the quaternary structural changes have not been evaluated. Intriguingly, the flavin reductases of two-component systems contain π- helices located at the tetramer interface. π-Helices are generated by a single amino acid insertion in an established α-helix to confer an evolutionary advantage. -

Structure-Function Relationship of Flavoproteins with Special Reference to P-Hydroxybenzoate Hydroxylase from Pseudomonas Fluorescens
Structure-function relationship of flavoproteins With special reference to p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens 0000 0330 0957 Promotor: dr. F. Müller, oud-hoogleraar in de Biochemie *j o^2ül, \Qrh> JU W.J . H. van Berkel Structure-function relationship of flavoproteins With special reference to p-hydroxybenzoate hydroxylase from Pseudomonasfluorescens Proefschrift ter verkrijging van de graad van doctor in de landbouwwetenschappen, op gezag van de rector magnificus, dr. H. C. van der Plas, in het openbaar te verdedigen op dinsdag 9 mei 1989 des namiddags te vier uur in de aula van de Landbouwuniversiteit te Wageningen BIBLIOTHEEK LANDBOUWUiNIVERSITEI') WAGEMNGFÏV (ôVo2?*>\} IZfo STELLINGEN 1. De enzym-nomenclatuur van FAD-bevattende monooxygenases dient nodigherzie n enu p todat egebrach t teworden . - Enzyme Nomenclature (1984)IUB ,Academi c Press (USA). - Ditproefschrift . 2. Rajue t al.gebruike n tenonrecht e deresultaten ,verkrege n inhoofdstu k 5va ndi t proefschrift,al sargumen t omaa nt e gevenda t cysteine residuen eenessentiël ero l spelen ind e katalyse van 4-hydroxyphenylacetaat-3-hydroxylase uit Pseudomonas putida. - Raju,S.G. , Kamath,A.V . and Vaidyanathan,CS . (1988) Bioch.Biophys .Res .Commun . 154,537-543 . 3. Het is jammer te moeten constateren dat "de fabrikantva n de beste biochemicaliën" anno 1989 de ratio lipoamide: diaphorase activiteit van lipoamide dehydrogenase slechts omschrijft alsee nmaa tvoo rd edenaturati e vanhe tenzym . -Massey ,V . and Veeger,C . (1961)Biochim . Biophys.Act a 48, 33-47. - Sigma catalogus (1989). 4. Zolang eenreactie-intermediai r in de natuurlijke reactie gekatalyseerd door p-hydroxybenzoaat hydroxylase nietword t gekarakteriseerd, laat staan waargenomen, blijft zijn voorkomen speculatief.