The Human Flavoproteome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ferric Reductase Activity of the Arsh Protein from Acidithiobacillus Ferrooxidans
J. Microbiol. Biotechnol. (2011), 21(5), 464–469 doi: 10.4014/jmb.1101.01020 First published online 13 April 2011 Ferric Reductase Activity of the ArsH Protein from Acidithiobacillus ferrooxidans Mo, Hongyu1,2, Qian Chen1,2, Juan Du1, Lin Tang1, Fang Qin1, Bo Miao2, Xueling Wu2, and Jia Zeng1,2* 1College of Biology, Hunan University, Changsha, Hunan 410082, P. R. China 2Department of Bioengineering, Central South University, Changsha, Hunan 410083, P. R. China Received: January 14, 2011 / Revised: March 10, 2011 / Accepted: March 11, 2011 The arsH gene is one of the arsenic resistance system in iron (free or chelated) into ferrous iron before its incorporation bacteria and eukaryotes. The ArsH protein was annotated into heme and nonheme iron-containing proteins. Ferric as a NADPH-dependent flavin mononucleotide (FMN) reductase catalyzes the reduction of complexed Fe3+ to reductase with unknown biological function. Here we complexed Fe2+ using NAD(P)H as the electron donor. The report for the first time that the ArsH protein showed resulting Fe2+ is subsequently released and incorporated high ferric reductase activity. Glu104 was an essential into iron-containing proteins [17]. residue for maintaining the stability of the FMN cofactor. Here we report for the first time that the ArsH protein The ArsH protein may perform an important role for showed high ferric reduction activity. The ArsH from A. cytosolic ferric iron assimilation in vivo. ferrooxidans may perform an important role as a NADPH- Keywords: Acidithiobacillus ferrooxidans, ArsH, flavoprotein, dependent ferric reductase for cytosolic ferric iron assimilation ferric reductase in vivo. MATERIALS AND METHODS Arsenic resistance genes are widespread in nature. -

Progressive Encephalopathy and Central Hypoventilation Related to Homozygosity of NDUFV1 Nuclear Gene, a Rare Mitochondrial Disease
Avens Publishing Group Inviting Innovations Open Access Case Report J Pediatr Child Care August 2019 Volume:5, Issue:1 © All rights are reserved by AL-Buali MJ, et al. AvensJournal Publishing of Group Inviting Innovations Progressive Encephalopathy Pediatrics & and Central Hypoventilation Child Care AL-Buali MJ*, Al Ramadhan S, Al Buali H, Al-Faraj J and Related to Homozygosity of Al Mohanna M Pediatric Department , Maternity Children Hospital , Saudi Arabia *Address for Correspondence: NDUFV1 Nuclear Gene, a Rare Al-buali MJ, Pediatric Consultant and Consultant of Medical Genetics, Deputy Chairman of Medical Genetic Unite, Pediatrics Department , Maternity Children Hospital, Al-hassa, Hofuf city, Mitochondrial Disease Saudi Arabia; E-mail: [email protected] Submission: 15 July 2019 Accepted: 5 August 2019 Keywords: Progressive encephalopathy; Central hypoventilation; Published: 9 August 2019 Nuclear mitochondrial disease; NDUFV1 gene Copyright: © 2019 AL-Buali MJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which Abstract permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Background: Mitochondrial diseases are a group of disorders caused by dysfunctional organelles that generate energy for our body. Mitochondria small double-membrane organelles found in of the most common groups of genetic diseases with a minimum every cell of the human body except red blood cells. Mitochondrial diseases are sometimes caused by mutations in the mitochondrial DNA prevalence of greater than 1 in 5000 in adults. Mitochondrial diseases that affect mitochondrial function. Other mitochondrial diseases are can be present at birth but can be manifested also at any age [2]. -

Expression of a Gene Encoding Acetolactate Synthase from Rice Complements Two Ilvh Mutants in Escherichia Coli
AJCS 4(6):430-436 (2010) ISSN:1835-2707 Expression of a gene encoding acetolactate synthase from rice complements two ilvH mutants in Escherichia coli Md. Shafiqul Islam Sikdar1, Jung-Sup Kim2* 1Faculty of Biotechnology, Jeju National University, Jeju, 690-756, Korea and Department of Agronomy, Hajee Mohammad Danesh Science and Technology University, Dinajpur-5200, Bangladesh 2Faculty of Biotechnology, Jeju National University, Jeju, 690-756, Korea *Corresponding Author: [email protected] Abstract Acetolactate synthase (ALS) is a thiamine diphosphate-dependent enzyme in the biosynthetic pathway leading to isoleucine, valine and leucine in plants. ALS is the target of several classes of herbicides that are effective to protect a broad range of crops. In this study, we describe the functional analysis of a gene encoding for ALS from rice (OsALS). Sequence analysis of an EST from rice revealed that it harbors a full-length open reading frame for OsALS encoding a protein of approximately 69.4 kDa and the N-terminal of OsALS contains a feature of chloroplast transit peptide. The predicted amino acid sequence of OsALS is highly homologous to those of weed ALSs among plant ALSs. The OsALS expression showed that the gene was functionally capable of complementing the two ilvH mutant strains of Escherichia coli. These results indicate that the OsALS encodes for an enzyme in acetolactate synthase in rice. Keywords: acetolactate synthase, rice (Oryza sativa), sequence analysis, functional complementation, ilvH mutants Abbreviations: ALS_Acetolactate synthase; BCAAs_Branched chain amino acids; Ile_Isoleucine; Val_Valine; Leu_Leucine; TPP_Thiamine diphosphate; CGSC_E. coli Genetic Stock Center; RGRC _Rice Genome Resource Center; ORF_Open reading frame; PCR_Polymerase chain reaction; Amp_Ampicillin; MM_M9 minimal medium; IPTG_Isopropyl β-D-thiogalactopyranoside. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

In Vitro Treatment of Hepg2 Cells with Saturated Fatty Acids Reproduces
© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 183-191 doi:10.1242/dmm.018234 RESEARCH ARTICLE In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis Inmaculada García-Ruiz1,*, Pablo Solís-Muñoz2, Daniel Fernández-Moreira3, Teresa Muñoz-Yagüe1 and José A. Solís-Herruzo1 ABSTRACT INTRODUCTION Activity of the oxidative phosphorylation system (OXPHOS) is Nonalcoholic fatty liver disease (NAFLD) represents a spectrum of decreased in humans and mice with nonalcoholic steatohepatitis. liver diseases extending from pure fatty liver through nonalcoholic Nitro-oxidative stress seems to be involved in its pathogenesis. The steatohepatitis (NASH) to cirrhosis and hepatocarcinoma that occurs aim of this study was to determine whether fatty acids are implicated in individuals who do not consume a significant amount of alcohol in the pathogenesis of this mitochondrial defect. In HepG2 cells, we (Matteoni et al., 1999). Although the pathogenesis of NAFLD analyzed the effect of saturated (palmitic and stearic acids) and remains undefined, the so-called ‘two hits’ model of pathogenesis monounsaturated (oleic acid) fatty acids on: OXPHOS activity; levels has been proposed (Day and James, 1998). Whereas the ‘first hit’ of protein expression of OXPHOS complexes and their subunits; gene involves the accumulation of fat in the liver, the ‘second hit’ expression and half-life of OXPHOS complexes; nitro-oxidative stress; includes oxidative stress resulting in inflammation, stellate cell and NADPH oxidase gene expression and activity. We also studied the activation, fibrogenesis and progression of NAFLD to NASH effects of inhibiting or silencing NADPH oxidase on the palmitic-acid- (Chitturi and Farrell, 2001). -

Targeting Glioblastoma Stem Cells Through Disruption of the Circadian Clock
Published OnlineFirst August 27, 2019; DOI: 10.1158/2159-8290.CD-19-0215 RESEARCH ARTICLE Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock Zhen Dong1, Guoxin Zhang1, Meng Qu2, Ryan C. Gimple1,3, Qiulian Wu1, Zhixin Qiu1, Briana C. Prager1,3, Xiuxing Wang1, Leo J.Y. Kim1,3, Andrew R. Morton3, Deobrat Dixit1, Wenchao Zhou4, Haidong Huang4, Bin Li5, Zhe Zhu1, Shideng Bao4, Stephen C. Mack6, Lukas Chavez7, Steve A. Kay2, and Jeremy N. Rich1 Downloaded from cancerdiscovery.aacrjournals.org on September 24, 2021. © 2019 American Association for Cancer Research. Published OnlineFirst August 27, 2019; DOI: 10.1158/2159-8290.CD-19-0215 ABSTRACT Glioblastomas are highly lethal cancers, containing self-renewing glioblastoma stem cells (GSC). Here, we show that GSCs, differentiated glioblastoma cells (DGC), and nonmalignant brain cultures all displayed robust circadian rhythms, yet GSCs alone displayed exquisite dependence on core clock transcription factors, BMAL1 and CLOCK, for optimal cell growth. Downregulation of BMAL1 or CLOCK in GSCs induced cell-cycle arrest and apoptosis. Chromatin immu- noprecipitation revealed that BMAL1 preferentially bound metabolic genes and was associated with active chromatin regions in GSCs compared with neural stem cells. Targeting BMAL1 or CLOCK attenu- ated mitochondrial metabolic function and reduced expression of tricarboxylic acid cycle enzymes. Small-molecule agonists of two independent BMAL1–CLOCK negative regulators, the cryptochromes and REV-ERBs, downregulated stem cell factors and reduced GSC growth. Combination of cryp- tochrome and REV-ERB agonists induced synergistic antitumor effi cacy. Collectively, these fi ndings show that GSCs co-opt circadian regulators beyond canonical circadian circuitry to promote stemness maintenance and metabolism, offering novel therapeutic paradigms. -
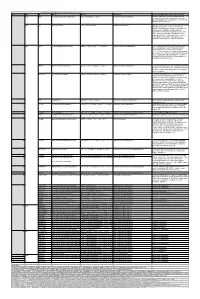
When the Reaction Is
Table S3. iJL1678-ME model modification (blocked reactions) Iter. Cat. ID Name Formula Subsystem Comments (When the reaction is turned on) 1 bp2 EDD 6-phosphogluconate dehydratase 6pgc_c⇌2ddg6p_c + h2o_c Pentose Phosphate Pathway Create a major effect of steep acetate overflow elevation in high growth. Comparing to the main glycolytic pathway, it is metabolicly less efficient but proteomicly more efficient. bp1 ICL Isocitrate lyase icit_c→glx_c + succ_c Anaplerotic Reactions Bypass for the main TCA cycle pathways from turning isocitrate to succinate, when ICL is turned on, Isocitrate dehydrogenase(ICDHyr), 2-Oxogluterate dehydrogenase(AKGDH) and Succinyl-CoA synthetase (ATP-forming,SUCOAS) would reduce. Ref. (1) and (2) shows that this reaction is off in higher growth. Ref. (3) shows that this reaction is converging to being off when the dynamic of respiration using enzyme kinetics is simulated. 2 bp1 ABTA 4-aminobutyrate transaminase 4abut_c + akg_c⇌glu__L_c + sucsal_c Arginine and Proline Metabolism Another backup pathway of succinate production, from 2-Oxoglutarate (akg). Respiration would be induced when it is on, since the flux through ETC(CYTBO3_4pp and ATPS4rpp) would increase. As it requires the co-factor pyridoxal 5'-phosphate(2−) to get catalyzed(4), indicating that this reaction is regulated by the flux of other reactions(pyridoxal 5'- phosphate(2-) production, etc.). 3 GLYAT Glycine C-acetyltransferase accoa_c + gly_c⇌2aobut_c + coa_c Glycine and Serine Metabolism A reaction that back up for the respiration. Reactions fluxes in TCA cycle would drop when this reaction is turned on. It also requires pyridoxal 5'-phosphate(2−) for the regulation. 4 NADTRHD NAD transhydrogenase nad_c + nadph_c⇌nadh_c + nadp_c Oxidative Phosphorylation A reaction that would make the transition between NAD and NADP metabolically more efficient. -
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Anamika Kothari Quang Ong Ron Caspi An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Peter D Karp Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Csac1394711Cyc: Candidatus Saccharibacteria bacterium RAAC3_TM7_1 Cellular Overview Connections between pathways are omitted for legibility. Tim Holland TM7C00001G0420 TM7C00001G0109 TM7C00001G0953 TM7C00001G0666 TM7C00001G0203 TM7C00001G0886 TM7C00001G0113 TM7C00001G0247 TM7C00001G0735 TM7C00001G0001 TM7C00001G0509 TM7C00001G0264 TM7C00001G0176 TM7C00001G0342 TM7C00001G0055 TM7C00001G0120 TM7C00001G0642 TM7C00001G0837 TM7C00001G0101 TM7C00001G0559 TM7C00001G0810 TM7C00001G0656 TM7C00001G0180 TM7C00001G0742 TM7C00001G0128 TM7C00001G0831 TM7C00001G0517 TM7C00001G0238 TM7C00001G0079 TM7C00001G0111 TM7C00001G0961 TM7C00001G0743 TM7C00001G0893 TM7C00001G0630 TM7C00001G0360 TM7C00001G0616 TM7C00001G0162 TM7C00001G0006 TM7C00001G0365 TM7C00001G0596 TM7C00001G0141 TM7C00001G0689 TM7C00001G0273 TM7C00001G0126 TM7C00001G0717 TM7C00001G0110 TM7C00001G0278 TM7C00001G0734 TM7C00001G0444 TM7C00001G0019 TM7C00001G0381 TM7C00001G0874 TM7C00001G0318 TM7C00001G0451 TM7C00001G0306 TM7C00001G0928 TM7C00001G0622 TM7C00001G0150 TM7C00001G0439 TM7C00001G0233 TM7C00001G0462 TM7C00001G0421 TM7C00001G0220 TM7C00001G0276 TM7C00001G0054 TM7C00001G0419 TM7C00001G0252 TM7C00001G0592 TM7C00001G0628 TM7C00001G0200 TM7C00001G0709 TM7C00001G0025 TM7C00001G0846 TM7C00001G0163 TM7C00001G0142 TM7C00001G0895 TM7C00001G0930 Detoxification Carbohydrate Biosynthesis DNA combined with a 2'- di-trans,octa-cis a 2'- Amino Acid Degradation an L-methionyl- TM7C00001G0190 superpathway of pyrimidine deoxyribonucleotides de novo biosynthesis (E. -

The Human Flavoproteome
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Archives of Biochemistry and Biophysics 535 (2013) 150–162 Contents lists available at SciVerse ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi Review The human flavoproteome ⇑ Wolf-Dieter Lienhart, Venugopal Gudipati, Peter Macheroux Graz University of Technology, Institute of Biochemistry, Petersgasse 12, A-8010 Graz, Austria article info abstract Article history: Vitamin B2 (riboflavin) is an essential dietary compound used for the enzymatic biosynthesis of FMN and Received 17 December 2012 FAD. The human genome contains 90 genes encoding for flavin-dependent proteins, six for riboflavin and in revised form 21 February 2013 uptake and transformation into the active coenzymes FMN and FAD as well as two for the reduction to Available online 15 March 2013 the dihydroflavin form. Flavoproteins utilize either FMN (16%) or FAD (84%) while five human flavoen- zymes have a requirement for both FMN and FAD. The majority of flavin-dependent enzymes catalyze Keywords: oxidation–reduction processes in primary metabolic pathways such as the citric acid cycle, b-oxidation Coenzyme A and degradation of amino acids. Ten flavoproteins occur as isozymes and assume special functions in Coenzyme Q the human organism. Two thirds of flavin-dependent proteins are associated with disorders caused by Folate Heme allelic variants affecting protein function. Flavin-dependent proteins also play an important role in the Pyridoxal 50-phosphate biosynthesis of other essential cofactors and hormones such as coenzyme A, coenzyme Q, heme, pyri- Steroids doxal 50-phosphate, steroids and thyroxine. Moreover, they are important for the regulation of folate Thyroxine metabolites by using tetrahydrofolate as cosubstrate in choline degradation, reduction of N-5.10-meth- Vitamins ylenetetrahydrofolate to N-5-methyltetrahydrofolate and maintenance of the catalytically competent form of methionine synthase. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The Effect of Increased Plastid Transketolase Activity on Thiamine
The effect of increased plastid transketolase activity on thiamine metabolism in transgenic tobacco plants Stuart James Fisk A thesis submitted for the degree of Doctor of Philosophy School of Biological Sciences University of Essex November 2015 I Acknowledgements I'd like to thank my supervisors Professor Christine Raines and Dr Tracy Lawson for all of the support and encouragement, Dr Julie Lloyd for the comments and suggestions given at board meetings and the University of Essex for funding this research. An extremely big thank you to all the members of plant group both past and present who have provided lots of help and technical support as well as plenty of laughs and an addiction to caffeine. But the biggest thanks of all go to my wife and best friend Alison Fisk. She always believed in me. II Summary Transketolase is a TPP dependent enzyme that affects the availability of intermediates in both the Calvin cycle and non-oxidative pentose phosphate pathway. Previous studies have indicated that changes to the activity level of transketolase can limit growth and development as well as the production of isoprenoids, starch, amino acids and thiamine. The overall aim of this project was to further advance the understanding of the mechanism linking increased TK activity and thiamine metabolism. Nicotiana tabacum mutants with increased total transketolase activity ~ 2 to 2.5 fold higher than WT plants were shown to have a reduced growth and chlorotic phenotype. In seedlings, these phenotypes were attributed to a reduction in seed thiamine content. Imbibition of TKox seeds in a thiamine solution produced plants that were comparable to WT plants. -

Oxidative Stress and Antioxidant Status in Hypo- and Hyperthyroidism
Chapter 8 Oxidative Stress and Antioxidant Status in Hypo- and Hyperthyroidism Mirela Petrulea, Adriana Muresan and Ileana Duncea Additional information is available at the end of the chapter http://dx.doi.org/10.5772/51018 1. Introduction Thyroid hormones are involved in the regulation of basal metabolic state and in oxidative metabolism [1]. They can cause many changes in the number and activity of mitochondrial respiratory chain components. This may result in the increased generation of reactive oxygen species (ROS) [2,3]. Oxidative stress is a general term used to describe a state of damage caused by ROS [4]. ROS have a high reactivity potential, therefore they are toxic and can lead to oxidative damage in cellular macromolecules such as proteins, lipids and DNA [5]. In fact, the cell contains a variety of substances capable of scavenging the free radicals, protecting them from harmful effects. Among the enzymatic antioxidants, are glutathione reductase (GR), glutathione peroxydase (GPx), catalase (CAT), superoxide dismutase (SOD), while the non-enzymatic antioxidants are glutathione (GSH), vitamin E, vitamin C, β- carotene, and flavonoids [6]. When ROS generation exceeds the antioxidant capacity of cells, oxidative stress develops [7]. Life means a continuous struggle for energy, which is required to fight against entropy. The most effective way to obtain energy is oxidation. Oxidative processes predominantly occur in mitochondria [8]. On the other hand, mitochondria are the favorite targets of thyroid hormones. During thyroid hormone synthesis, there is a constant production of oxygenated water, which is absolutely indispensable for iodine intrafollicular oxidation in the presence of thyroid peroxidase. In recent years, the possible correlation between impaired thyroid gland function and reactive oxygen species has been increasingly taken into consideration [9].