Selective Benzylic C–H Monooxygenation Mediated by Iodine Oxides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent Office Patented Apr
3,378,337 United States Patent Office Patented Apr. 16, 1968 1. 2 and the hydrogen peroxide. This result is achieved without 3,378,337 contaminating the reaction mixture. The final solution PREPARATHON OF ODEC ACID AND DERVATIVES THEREOF contains substantially pure iodic acid, and can be evap Ricardo O. Bach, Gastonia, N.C., assignor to Lithium orated and dehydrated to obtain the iodic acid, or the Corporation of America, Inc., New York, N.Y., a cor anhydride thereof, or it can be used as a medium for the poration of Minnesota direct production of salts of iodic acid. No Drawing. Fied May 17, 1965, Ser. No. 456,561 In carrying out the method of the present invention, 12 Claims. (CI. 23-85) the iodic acid solution employed in forming the reaction mixture should contain sufficient iodic acid to provide a O hydrogen ion and iodate ion concentration in the reaction ABSTRACT OF THE DISCLOSURE mixture which will favor oxidation of iodine to its penta A method of preparing solutions of substantially pure valent state and impede decomposition of hydrogen iodic acid from which salts of the acid, the anhydride peroxide. Generally speaking, the quantity of iodic acid thereof and/or the crystalline form of the acid can be present in the starting solution should not be below about 5 0.5%, with especially good results being attained with directly obtained. The method involves reacting iodine from about 1% to about 10%, usually about 5%, of the with hydrogen peroxide in the presence of a relatively quantity of iodic acid to be produced. -

Crown Chemical Resistance Chart
Crown Polymers, Corp. 11111 Kiley Drive Huntley, IL. 60142 USA www.crownpolymers.com 847-659-0300 phone 847-659-0310 facisimile 888-732-1270 toll free Chemical Resistance Chart Crown Polymers Floor and Secondary Containment Systems Products: CrownShield covers the following five (4) formulas: CrownShield 50, Product No. 320 CrownCote, Product No. 401 CrownShield 40-2, Product No. 323 CrownShield 28, Product No. 322 CrownPro AcidShield, Product No. 350 CrownCote AcidShield, Product No. 430 CrownPro SolventShield, Product No. 351 CrownCote SolventShield, Product No. 440 This chart shows chemical resistance of Crown Polymers foundational floor and secondary containment product line that would be exposed to chemical spill or immersion conditions. The chart was designed to provide general product information. For specific applications, contact your local Crown Polymers Floor and Secondary Containment Representative or call direct to the factory. ; Resistant to chemical immersion up to 7 days followed by wash down with water 6 Spillage environments that will be cleaned up within 72 hours after initial exposure. 9 Not Recommended Chemical CrownShield SolventShield AcidShield Chemical CrownShield SolventShield AcidShield 1, 4-Dichloro-2-butene 9 6 6 Aluminum Bromate ; ; ; 1, 4-Dioxane 9 6 6 Aluminum Bromide ; ; ; 1-1-1 Trichloroethane 9 ; ; Aluminum Chloride ; ; ; 2, 4-Pentanedione 6 ; 6 Aluminum Fluoride (25%) ; ; ; 3, 4-Dichloro-1-butene 6 6 6 Aluminum Hydroxide ; ; ; 4-Picoline (0-50%) 9 6 6 Aluminum Iodine ; ; ; Acetic Acid (0-15%) 9 6 6 -
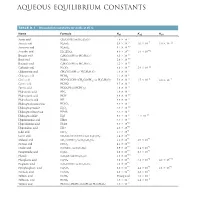
Aqueous Equilibrium Constants
BLB_APP_D_1062-1063hr.qxp 11/8/10 2:27 PM Page 1062 APPENDIX D AQUEOUS EQUILIBRIUM CONSTANTS TABLE D.1 • Dissociation Constants for Acids at 25 ˚C Name Formula Ka1 Ka2 Ka3 -5 Acetic acid CH3COOH (or HC2H3O2) 1.8 * 10 * -3 * -7 * -12 Arsenic acid H3AsO4 5.6 10 1.0 10 3.0 10 * -10 Arsenous acid H3AsO3 5.1 10 * -5 * -12 Ascorbic acid H2C6H6O6 8.0 10 1.6 10 * -5 Benzoic acid C6H5COOH (or HC7H5O2) 6.3 10 * -10 Boric acid H3BO3 5.8 10 * -5 Butanoic acid C3H7COOH (or HC4H7O2) 1.5 10 * -7 * -11 Carbonic acid H2CO3 4.3 10 5.6 10 * -3 Chloroacetic acid CH2ClCOOH (or HC2H2O2Cl) 1.4 10 * -2 Chlorous acid HClO2 1.1 10 * -4 * -5 -7 Citric acid HOOCC(OH) (CH2COOH)2 (or H3C6H5O7) 7.4 10 1.7 10 4.0 * 10 - Cyanic acid HCNO 3.5 * 10 4 * -4 Formic acid HCOOH (or HCHO2) 1.8 10 * -5 Hydroazoic acid HN3 1.9 10 - Hydrocyanic acid HCN 4.9 * 10 10 - Hydrofluoric acid HF 6.8 * 10 4 - -7 Hydrogen chromate ion HCrO4 3.0 * 10 * -12 Hydrogen peroxide H2O2 2.4 10 - -2 Hydrogen selenate ion HSeO4 2.2 * 10 * -8 * -19 Hydrogen sulfide H2S 9.5 10 1 10 - Hypobromous acid HBrO 2.5 * 10 9 - Hypochlorous acid HClO 3.0 * 10 8 - Hypoiodous acid HIO 2.3 * 10 11 * -1 Iodic acid HIO3 1.7 10 * -4 Lactic acid CH3CH(OH)COOH (or HC3H5O3) 1.4 10 * -3 * -6 Malonic acid CH2(COOH)2 (or H2C3H2O4) 1.5 10 2.0 10 * -4 Nitrous acid HNO2 4.5 10 * -2 * -5 Oxalic acid (COOH)2 (or H2C2O4) 5.9 10 6.4 10 * -2 * -9 Paraperiodic acid H5IO6 2.8 10 5.3 10 * -10 Phenol C6H5OH (or HC6H5O) 1.3 10 * -3 * -8 * -13 Phosphoric acid H3PO4 7.5 10 6.2 10 4.2 10 * -5 Propionic acid C2H5COOH (or HC3H5O2) 1.3 10 * -

APPENDIX G Acid Dissociation Constants
harxxxxx_App-G.qxd 3/8/10 1:34 PM Page AP11 APPENDIX G Acid Dissociation Constants § ϭ 0.1 M 0 ؍ (Ionic strength ( † ‡ † Name Structure* pKa Ka pKa ϫ Ϫ5 Acetic acid CH3CO2H 4.756 1.75 10 4.56 (ethanoic acid) N ϩ H3 ϫ Ϫ3 Alanine CHCH3 2.344 (CO2H) 4.53 10 2.33 ϫ Ϫ10 9.868 (NH3) 1.36 10 9.71 CO2H ϩ Ϫ5 Aminobenzene NH3 4.601 2.51 ϫ 10 4.64 (aniline) ϪO SNϩ Ϫ4 4-Aminobenzenesulfonic acid 3 H3 3.232 5.86 ϫ 10 3.01 (sulfanilic acid) ϩ NH3 ϫ Ϫ3 2-Aminobenzoic acid 2.08 (CO2H) 8.3 10 2.01 ϫ Ϫ5 (anthranilic acid) 4.96 (NH3) 1.10 10 4.78 CO2H ϩ 2-Aminoethanethiol HSCH2CH2NH3 —— 8.21 (SH) (2-mercaptoethylamine) —— 10.73 (NH3) ϩ ϫ Ϫ10 2-Aminoethanol HOCH2CH2NH3 9.498 3.18 10 9.52 (ethanolamine) O H ϫ Ϫ5 4.70 (NH3) (20°) 2.0 10 4.74 2-Aminophenol Ϫ 9.97 (OH) (20°) 1.05 ϫ 10 10 9.87 ϩ NH3 ϩ ϫ Ϫ10 Ammonia NH4 9.245 5.69 10 9.26 N ϩ H3 N ϩ H2 ϫ Ϫ2 1.823 (CO2H) 1.50 10 2.03 CHCH CH CH NHC ϫ Ϫ9 Arginine 2 2 2 8.991 (NH3) 1.02 10 9.00 NH —— (NH2) —— (12.1) CO2H 2 O Ϫ 2.24 5.8 ϫ 10 3 2.15 Ϫ Arsenic acid HO As OH 6.96 1.10 ϫ 10 7 6.65 Ϫ (hydrogen arsenate) (11.50) 3.2 ϫ 10 12 (11.18) OH ϫ Ϫ10 Arsenious acid As(OH)3 9.29 5.1 10 9.14 (hydrogen arsenite) N ϩ O H3 Asparagine CHCH2CNH2 —— —— 2.16 (CO2H) —— —— 8.73 (NH3) CO2H *Each acid is written in its protonated form. -

IODINE Its Properties and Technical Applications
IODINE Its Properties and Technical Applications CHILEAN IODINE EDUCATIONAL BUREAU, INC. 120 Broadway, New York 5, New York IODINE Its Properties and Technical Applications ¡¡iiHiüíiüüiütitittüHiiUitítHiiiittiíU CHILEAN IODINE EDUCATIONAL BUREAU, INC. 120 Broadway, New York 5, New York 1951 Copyright, 1951, by Chilean Iodine Educational Bureau, Inc. Printed in U.S.A. Contents Page Foreword v I—Chemistry of Iodine and Its Compounds 1 A Short History of Iodine 1 The Occurrence and Production of Iodine ....... 3 The Properties of Iodine 4 Solid Iodine 4 Liquid Iodine 5 Iodine Vapor and Gas 6 Chemical Properties 6 Inorganic Compounds of Iodine 8 Compounds of Electropositive Iodine 8 Compounds with Other Halogens 8 The Polyhalides 9 Hydrogen Iodide 1,0 Inorganic Iodides 10 Physical Properties 10 Chemical Properties 12 Complex Iodides .13 The Oxides of Iodine . 14 Iodic Acid and the Iodates 15 Periodic Acid and the Periodates 15 Reactions of Iodine and Its Inorganic Compounds With Organic Compounds 17 Iodine . 17 Iodine Halides 18 Hydrogen Iodide 19 Inorganic Iodides 19 Periodic and Iodic Acids 21 The Organic Iodo Compounds 22 Organic Compounds of Polyvalent Iodine 25 The lodoso Compounds 25 The Iodoxy Compounds 26 The Iodyl Compounds 26 The Iodonium Salts 27 Heterocyclic Iodine Compounds 30 Bibliography 31 II—Applications of Iodine and Its Compounds 35 Iodine in Organic Chemistry 35 Iodine and Its Compounds at Catalysts 35 Exchange Catalysis 35 Halogenation 38 Isomerization 38 Dehydration 39 III Page Acylation 41 Carbón Monoxide (and Nitric Oxide) Additions ... 42 Reactions with Oxygen 42 Homogeneous Pyrolysis 43 Iodine as an Inhibitor 44 Other Applications 44 Iodine and Its Compounds as Process Reagents ... -

Chemical Resistance 100% SOLIDS EPOXY SYSTEMS
Chemical Resistance 100% SOLIDS EPOXY SYSTEMS CHEMICAL 8300 SYSTEM 8200 SYSTEM 8000 SYSTEM OVERKOTE PLUS HD OVERKOTE HD OVERKRETE HD BASED ON ONE YEAR IMMERSION TESTING –––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––– Acetic Acid (0-15%) G II Acetonitrile LLG L Continuous Immersion Acetone (0-20%) LLL Acetone (20-30%) Suitable for continuous immersion in that chemical (based on LLG Acetone (30-50%) L G I ONE YEAR testing) to assure unlimited service life. Acetone (50-100%) G II Acrylamide (0-50%) LLL G Short-Term Exposure Adipic Acid Solution LLL Alcohol, Isopropyl LLL Suitable for short-term exposure to that chemical such as Alcohol, Ethyl LLG secondary containment (72 hours) or splash and spill Alcohol, Methyl LLI (immediate clean-up). Allyl Chloride LLI Allylamine (0-20%) L L I Allylamine (20-30%) L G I I Not Suitable Allylamine (30-50%) GGI Not suitable for any exposure to that chemical. Aluminum Bromide LL– Aluminum Chloride L L – Aluminum Fluoride (0-25%) L L – This chart shows chemical resistance of our various Aluminum Hydroxide LLL 1 topping materials (90 mils – ⁄4"). These ratings are based on Aluminum Iodide LL– temperatures being ambient. At higher temperatures, chemical Aluminum Nitrate LL– resistance may be effected. When chemical exposure is Aluminum Sodium Chloride L L – minimal to non-existent, a 9000 System–FlorClad™ HD or Aluminum Sulfate LLL 4600 System– BriteCast™ HD may be used. Alums L L L 2-Aminoethoxyethanol Resistance data is listed with the assumption that the material GGG has properly cured for at least four days, at recommended Ammonia – Wet L L – temperatures, prior to any chemical exposure. -
Thermal-Chem Corp. High Performance Epoxy Topcoats Products
Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E56 E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re DecoThane ArmorBond SolventGard DecoTop P92 Chemical 750 755 757 728 731 736 705 748 1056 1060 Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E56 E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re DecoThane ArmorBond SolventGard DecoTop P92 Chemical 750 755 757 728 731 736 705 748 1056 1060 1, 4-Dichloro-2-butene U E E U U U U U U U 1, 4-Dioxane U < < U U U U U U U 1-1-1 Trichloroethane E E E F F G G F G G 2, 4-Pentanedione F E E < < < < < F F 3, 4-Dichloro-1-butene < E E U U U U U U U 4-Picoline (0-50%) U < < U U U U U U V Acetic Acid (0-5%) E E E G G G G G G G Acetic Acid (5-15%) < E E < < < < < F F Acetone(0-20%) E E E F G G G G E F Acetone (20-30% < E E < < < < < < < Acetone (30-50%) < E E < < < < < < < Acetone (50-100%) < F F < < < < < E < Acetonitrile F E E < < < < < < < Acrylamide (0-50%) E E E F G G G G E F Adipic Acid Solution E E E E E E E E E G Alcohol, Ethyl E E E F F F F F G F Alcohol, Isopropyl E E E F F F F F G F Alcohol, Methyl < E E < < < < < < < Allyl Chloride < E E U U U U U < < Allylamine (0-20%) < E E U U U U U < < Allylamine (20-30%) < E E U U U U U < < Allylamine (30-50%) U < < U U U U U U U Aluminum Bromate E E E F G G G G E E Aluminum Bromide E E E E E E E E E E Aluminum Chloride E E E E E E E E E E Aluminum Fluoride (25%) E E E E E E E E E E Polyurethane Thermal-Chem Corp. -

BOSC Review of US EPA ORD Research Programs
May 8, 2017 Robert Kavlock, Ph.D. Acting Assistant Administrator Office of Research and Development U.S. Environmental Protection Agency Dear Dr. Kavlock: On behalf of the Board of Scientific Counselors (BOSC), I am pleased to provide you a collection of reports addressing Charge Questions posed by five of the National Research Program areas and the four cross- cutting Roadmap programs. In general, we have found these programs to be on track to meet the objectives in their current Strategic Research Action Plans (StRAPs) and Roadmaps. We provide a series of recommendations to continue to strengthen the excellent research being done in ORD, and look forward to working with you in the future on these programs. Sincerely, Deborah L. Swackhamer, Ph.D. Chair, BOSC Cc: Bruce Rodan, Associate Director of Science REVIEW OF U.S. EPA OFFICE OF RESEARCH AND DEVELOPMENT’S RESEARCH PROGRAMS BOSC EXECUTIVE COMMITTEE Chair Deborah L. Swackhamer, Ph.D. James N. Galloway, Ph.D. I. Leslie Rubin, M.D. University of Minnesota University of Virginia Developmental Pediatric Specialists Viney Aneja, Ph.D. Earthea A. Nance, Ph.D., P.E. Sandra Smith, M.S. North Carolina State University Texas Southern University AECOM Shahid Chaudhry Paula Olsiewski, Ph.D., P.E. Gina Solomon, M.D., M.P.H. California Energy Commission Alfred P. Sloan Foundation California EPA Elizabeth Corley, Ph.D. Diane E. Pataki, Ph.D. Ponisseril Somasundaran, Ph.D. Arizona State University The University of Utah Columbia University Susan E. Cozzens, Ph.D. Robert Richardson, Ph.D. Tammy P. Taylor, Ph.D. Georgia Institute of Technology Michigan State University Pacific Northwest National Laboratory Courtney Flint, Ph.D. -

General Chemical Resistance of a Fluoroelastomer Coatings & Sealants Fluoroelastomer
General Chemical Resistance of a Fluoroelastomer Coatings & Sealants Fluoroelastomer www.pelseal.com Pelseal Technologies’ state-of-the-art fluid coatings, surface area to volume. Therefore, the ratings in the caulks, adhesives and sealants are manufactured from accompanying listing should be used only as a general fluoroelastomer rubber. Our products resist attack, to guide, not as specific recommendations. varying degrees, by a broad spectrum of industrial chemicals, fuels, oils, and other substances. While the information is presented by us in good faith, no guarantee, expressed or implied, can be given regarding We have researched the effect of the listed chemicals the accuracy of these ratings. on fluoroelastomers at various temperatures, concentrations, and exposure times. Our information is based on extensive In those cases where no data are available, the rating laboratory tests and experiments that have been performed shown is the considered, conditional opinion of experienced on fluoroelastomers themselves by a number of outside rubber chemists and compounders. organizations, including manufacturers, end users, suppliers, and trade associations. We strongly urge you to test our products under actual or simulated service conditions, before you purchase them, The information is believed to be reliable. However, the rather than assume that they will perform satisfactorily in degree of fluid resistance of an elastomer to deterioration your specific application. In order to assist you in learning by a specific substance is dependent on many variables, more about the chemical resistance of Pelseal's products including the concentration and temperature of the material; to the substance shown, the following rating code has the frequency and duration of exposure; the velocity of been used: flow and aeration; mechanical action; and the ratio of contact CODE E Excellent Resistance - little or minor attack by the material on fluoroelastomers; 0-5% volume swell where applicable. -

Chemical Resistance Chart
Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E56 E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re DecoThane ArmorBond SolventGard DecoTop P92 Chemical 750 755 757 728 731 736 705 748 1056 1060 Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re ThermalCrete ArmorBond SolventGard DecoFinish P81 Chemical 750 755 757 728 731 736 705 748 1030 1062 1, 4-Dichloro-2-butene U E E U U U U U U U 1, 4-Dioxane U < < U U U U U U U 1-1-1 Trichloroethane E E E F F G G F G G < < < < < 2, 4-Pentanedione F E E F F 3, 4-Dichloro-1-butene < E E U U U U U U U 4-Picoline (0-50%) U < < U U U U U V V Acetic Acid (0-5%) E E E G G G G G E G < < < < < < Acetic Acid (5-15%) E E E F Acetone(0-20%) E E E F G G G G E F Acetone (20-30% < E E < < < < < E < Acetone (30-50%) < E E < < < < < E < Acetone (50-100%) < F F < < < < < E < Acetonitrile F E E < < < < < E < Acrylamide (0-50%) E E E F G G G G F Adipic Acid Solution E E E E E E E E G Alcohol, Ethyl E E E F F F F F F Alcohol, Isopropyl E E E F F F F F G F Alcohol, Methyl < E E < < < < < < < Allyl Chloride < E E U U U U U E < Allylamine (0-20%) < E E U U U U U < Allylamine (20-30%) < E E U U U U U < Allylamine (30-50%) U < < U U U U U U Aluminum Bromate E E E F G G G G E Aluminum Bromide E E E E E E E E E Aluminum Chloride E E E E E E E E E Aluminum Fluoride (25%) E E E E E E E E E Polyurethane Thermal-Chem Corp. -

Photolysis of Frozen Iodate Salts As a Source of Active Iodine in the Polar Environment
Atmos. Chem. Phys., 16, 12703–12713, 2016 www.atmos-chem-phys.net/16/12703/2016/ doi:10.5194/acp-16-12703-2016 © Author(s) 2016. CC Attribution 3.0 License. Photolysis of frozen iodate salts as a source of active iodine in the polar environment Óscar Gálvez1,a, M. Teresa Baeza-Romero2, Mikel Sanz2,b, and Alfonso Saiz-Lopez3 1Departamento de Física Molecular, Instituto de Estructura de la Materia, IEM-CSIC, 28006 Madrid, Spain 2Escuela de Ingeniería Industrial, Universidad de Castilla-La Mancha, 45071 Toledo, Spain 3Department of Atmospheric Chemistry and Climate, Institute of Physical Chemistry Rocasolano, CSIC, 28006 Madrid, Spain anow at: Departamento de Física Interdisciplinar, Facultad de Ciencias, Universidad Nacional de Educación a Distancia, 28040 Madrid, Spain bnow at: Institute of Physical Chemistry Rocasolano, CSIC, 28006 Madrid, Spain Correspondence to: Óscar Gálvez ([email protected], [email protected]) Received: 15 September 2015 – Published in Atmos. Chem. Phys. Discuss.: 15 October 2015 Revised: 22 September 2016 – Accepted: 26 September 2016 – Published: 12 October 2016 Abstract. Reactive halogens play a key role in the oxida- 1 Introduction tion capacity of the polar troposphere. However, sources and mechanisms, particularly those involving active iodine, are still poorly understood. In this paper, the photolysis of an Atmospheric iodine compounds are present in the marine atmospherically relevant frozen iodate salt has been experi- and polar boundary layers (Saiz-Lopez et al., 2012), where mentally studied using infrared (IR) spectroscopy. The sam- they play a relevant role in catalytic ozone destruction (Saiz- ples were generated at low temperatures in the presence of Lopez et al., 2007b; Read et al., 2008), and they could also different amounts of water. -

Stencil Remover 285 Safety Data Sheet 01 June 2015
Safety Data Sheet Section 1: Identification of the Substance/Mixture and of the Company/Undertaking 1.1 Product identifier Product Name • Stencil Remover 285 Product Description • Clear liquid. 1.2 Relevant identified uses of the substance or mixture and uses advised against Relevant identified use(s) • Liquid stencil remover concentrate 1.3 Details of the supplier of the safety data sheet Manufacturer • IKONICS Corporation 4832 Grand Ave. Duluth, MN 55807 United States www.ikonics.com [email protected] Telephone (General) • (218) 628-2217 Telephone (General) • (800) 328-4261 - Toll free 1.4 Emergency telephone number Chemtrec • 1-800-424-9300 - Within USA and Canada • +1 703-527-3887 - Outside USA and Canada (collect calls accepted) Section 2: Hazards Identification EU/EEC According to EU Directive 1272/2008 (CLP)/REACH 1907/2006 [amended by 453/2010] According to EU Directive 67/548/EEC (DSD) or 1999/45/EC (DPD) 2.1 Classification of the substance or mixture CLP • Oxidizing Liquids 2 - H272 Skin Corrosion 1 - H314 Serious Eye Damage 1 - H318 DSD/DPD • Corrosive (C) R35 2.2 Label Elements CLP DANGER Hazard statements • H272 - May intensify fire; oxidizer H314 - Causes severe skin burns and eye damage Stencil Remover 285 Safety Data Sheet Page 1 of 12 H318 - Causes serious eye damage Precautionary statements Prevention • P210 - Keep away from heat. P220 - Keep/Store away from clothing and other combustible materials. P221 - Take any precaution to avoid mixing with combustibles P260 - Do not breathe dusts or mists. P264 - Wash thoroughly after handling. P280 - Wear protective gloves/protective clothing/eye protection/face protection.