As a Platform Molecule for the Synthesis of Different
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Broda 1 Potential Synthesis Pathway for Redox-Active DNA Nucleoside
Broda 1 Potential Synthesis Pathway for Redox-Active DNA Nucleoside Nicole M. Broda Thesis Advisor: Robert Kuchta, Ph.D.: Department of Chemistry and Biochemistry Committee Members: Liu Xuedong, Ph.D.: Department of Chemistry and Biochemistry Levente Szentkirályi, Ph.D.: Program For Writing and Rhetoric Undergraduate Thesis March 23rd, 2018 University of Colorado at Boulder Department of Chemistry and Biochemistry Broda 2 Acknowledgements I am incredibly thankful for the mentorship and experience I have received from Robert Kuchta, who gave me the opportunity to be an undergraduate research assistant and supported me during numerous independent projects. Likewise, I want to extend my thanks to the other members of the Kuchta lab that have been crucial in my development as a researcher and in the intricacies of this thesis: Sarah Dickerson, for her guidance through the research process and creation of a friendly working environment; Ayman Alawneh, for his mentorship in the art of organic synthesis from the bottom-up and for answering my numerous questions about the theory behind it all; and Michelle Ledru, for sharing the long synthesis days and learning the process with me. Additionally, thank you to my honors committee members Liu Xuedong and Levente Szentkirályi for volunteering their time to participate in this undergraduate thesis defense. A special thanks to Levente Szentkirályi, for helping me through the thesis writing process from the draft to the final and for instilling in me the responsibility we as a scienctific community have to make science accessible to interdisciplinary readers and the general public. I extend my appreciation to Michael J. -

GRAS Notice 789 for Erythritol
GRAS Notice (GRN) No. 789 https://www.fda.gov/food/generally-recognized-safe-gras/gras-notice-inventory. Toi• Strategies ~~~~G~~[)) JUN 7 20'8 Innovative solutions Sound science OFFICE OF FOOD ADDITIVE SAFE1Y June 5, 2018 Dr. Dennis Keefe Director, Division of Biotechnology and GRAS Notice Review Office of Food Additive Safety (HFS-200) Center for Food Safety and Applied Nutrition Food and Drug Administration 5100 Paint Branch Parkway College Park, MD 20740-3835 Subject: GRAS Notification - Erythritol Dear Dr. Keefe: On behalf of Cargill, Incorporated, ToxStrategies, Inc. (its agent) is submitting, for FDA review, a copy of the GRAS notification as required. The enclosed document provides notice of a claim that the food ingredient, erythritol, described in the enclosed notification is exempt from the premarket approval requirement of the Federal Food, Drug, and Cosmetic Act because it has been determined to be generally recognized as safe (GRAS), based on scientific procedures, for addition to food. If you have any questions or require additional information, please do not hesitate to contact me at 630-352-0303, or [email protected]. Sincerely, (b) (6) Donald F. Schmitt, M.P.H. Senior Managing Scientist ToxStrategies, Inc., 931 W. 75th St. , Suite 137, PMB 263, Naperville, IL 60565 1 Office (630) 352-0303 • www.toxstrategies.com GRAS Determination of Erythritol for Use in Human Food JUNES,2018 Innovative solutions s ,..,.,',--.r-.r--.r--. OFFICE OF FOOD ADDITIVE SAFE1Y GRAS Determination of Erythritol for Use in Human Food SUBMITTED BY: Cargill, Incorporated 15407 McGinty Road West Wayzata, MN 55391 SUBMITTED TO: U.S. Food and Drug Administration Center for Food Safety and Applied Nutrition Office of Food Additive Safety HFS-200 5100 Paint Branch Parkway College Park MD 20740-3835 CONTACT FOR TECHNICAL OR OTIIER INFORMATION Donald F. -

“Polyols: a Primer for Dietetic Professionals” Is a Self-Study
1 “Polyols: A primer for dietetic professionals” is a self-study module produced by the Calorie Control Council, an accredited provider of continuing professional education (CPE) for dietetic professionals by the Commission on Dietetic Registration. It provides one hour of level 1 CPE credit for dietetic professionals. The full text of the module is in the notes section of each page, and is accompanied by summary points and/or visuals in the box at the top of the page. Directions for obtaining CPE are provided at the end of the module. 2 After completing this module, dietetic professionals will be able to: • Define polyols. • Identify the various types of polyols found in foods. • Understand the uses and health effects of polyols in foods. • Counsel clients on how to incorporate polyols into an overall healthful eating pattern. 3 4 Polyols are carbohydrates that are hydrogenated, meaning that a hydroxyl group replaces the aldehyde or ketone group found on sugars. Hydrogenated monosaccharides include erythritol, xylitol, sorbitol, and mannitol. Hydrogenated disaccharides include lactitol, isomalt, and maltitol. And hydrogenated starch hydrolysates (HSH), or polyglycitols (a wide range of corn syrups and maltodextrins), are formed from polysaccharides (Grabitske and Slavin 2008). 5 Nearly 54 percent of Americans are trying to lose weight, more than ever before. Increasingly, they are turning toward no- and low-sugar, and reduced calorie, foods and beverages to help them achieve their weight loss goals (78% of Americans who are trying to lose weight) (CCC 2010). Polyols, found in many of these foods, are becoming a subject of more interest. 6 They are incompletely digested , therefore are sometimes referred to as “low- digestible carbohydrates.” Polyols are not calorie free, as there is some degree of digestion and absorption of the carbohydrate. -

Catalyst in Basic Oleochemicals
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Diponegoro University Institutional Repository Bulletin of Chemical Reaction Engineering & Catalysis, 2(2-3), 2007, 22-31 Catalyst in Basic Oleochemicals Eva Suyenty, Herlina Sentosa, Mariani Agustine, Sandy Anwar, Abun Lie, and Erwin Sutanto * Research and Development Department, PT. Ecogreen Oleochemicals Jln. Pelabuhan Kav 1, Kabil, Batam 29435, Telp/Fax: (0778)711374 Presented at Symposium and Congress of MKICS 2007, 18-19 April 2007, Semarang, Indonesia Abstract Currently Indonesia is the world largest palm oil producer with production volume reaching 16 million tones per annum. The high crude oil and ethylene prices in the last 3 – 4 years contribute to the healthy demand growth for basic oleochemicals: fatty acids and fatty alcohols. Oleochemicals are starting to replace crude oil derived products in various applications. As widely practiced in petrochemical industry, catalyst plays a very important role in the production of basic oleochemicals. Catalytic reactions are abound in the production of oleochemicals: Nickel based catalysts are used in the hydrogenation of unsaturated fatty ac- ids; sodium methylate catalyst in the transesterification of triglycerides; sulfonic based polystyrene resin catalyst in esterification of fatty acids; and copper chromite/copper zinc catalyst in the high pressure hydro- genation of methyl esters or fatty acids to produce fatty alcohols. To maintain long catalyst life, it is crucial to ensure the absence of catalyst poisons and inhibitors in the feed. The preparation methods of nickel and copper chromite catalysts are as follows: precipitation, filtration, drying, and calcinations. Sodium methy- late is derived from direct reaction of sodium metal and methanol under inert gas. -

Study of the Partial and Exhaustive Intramolecular Dehydration of D-Sorbitol
Available online at http://journal-of-agroalimentary.ro Journal of Journal of Agroalimentary Processes and Agroalimentary Processes and Technologies 2013, 19(2), 259-270 Technologies Study of the partial and exhaustive intramolecular dehydration of D-sorbitol Ileana Cocan *, Monica Viorica Negrea, Ionel Vasile Jianu ”Dunărea de Jos” University of Galati, Faculty of Food Science and Engineering, Domnească Street, 47, RO-800008, Galati, Romania Received: 07 February 2013; Accepted: 09 March 2013 ______________________________________________________________________________________ Abstract Sweetener, humectants, sequestrant, texturizer, stabilizer, bulking agent, D-sorbitol represents a leading product in the processing of the polysorbate class. The scope of this work is to obtain details on the preparation and accession of D-sorbitol mono- and dianhydride as a method of chemical protection of primary and secondary hydroxyl functional groups (1:4) (3:6) as part of the strategy of controlled processing of polysorbates containing “homogeneous” polyoxyethylenic chains (n = 3, 6, 9, 18). This work is investigating the dependence of yield of internal dehydration (partial and/or exhaustive, direct of D-sorbitol) on temperature (100-160°C and 120-200°C , respectively ) and duration (5–35 minutes and 10–100 minutes , respectively ) for the processing of 1,4-sorbitan and 2,5-isosorbide. The evolution of the hydroxyl number (mg KOH/g D-sorbitol) (mg KOH/g 1,4-sorbitan) related to the theoretical (initial) value was followed and optimal values for the yield and dehydration parameters were established. Also in this work, the mathematical modeling of the dependence curves experimentally registered is realized. Keywords : izosorbide, D-sorbitol, D-glucitol, D-sorbit, 1,4-sorbitan ______________________________________________________________________________________ 1. -

Step-Growth Polymerisation of Alkyl Acrylates Via Concomitant Oxa-Michael and Transesterification Reactions
Step-growth polymerisation of alkyl acrylates via concomitant oxa-Michael and transesterification reactions Karin Ratzenböck,a David Pahovnikb and Christian Slugovc *a,c a Christian Doppler Laboratory for Organocatalysis in Polymerization, Stremayrgasse 9, A 8010 Graz, Austria b National Institute of Chemistry, Department of Polymer Chemistry and Technology, Hajdrihova 19, 1000 Ljubljana, Slovenia c Institute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, A 8010 Graz, Austria Herein we propose an auto-tandem catalytic approach towards the preparation of poly(ester- ether)s from simple alkyl acrylates and diols. By combining oxa-Michael addition with transesterification the preparation of hydroxy functionalized acrylate monomers can be avoided. Introduction. Polymerisation of acrylates is typically performed by free radical polymerisation. Formed poly(acrylates) based on C-C main chains are widely used as paintings, surface coatings, adhesives, or fibres.1 However, the oxa-Michael addition reaction2 presents an alternative method for the polymerisation of acrylates leading to potentially more easily degradable poly(ester-ether)s in which functional groups are incorporated in the polymer main chain. Oxa-Michael addition polymerisation of hydroxyalkyl acrylates, also referred to as hydrogen-transfer polymerisation, was first reported in 1975.3 The polymerisation of 2- hydroxyethyl acrylate (HEA) could be initiated with LiH or PPh3. Since then few studies on the polymerisation of hydroxy functionalized -
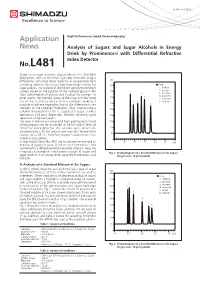
Analysis of Sugars and Sugar Alcohols in Energy Drink by Prominence-I with Differential Refreactive Index Detector
LAAN-A-LC-E258 Application High Performance Liquid Chromatography News Analysis of Sugars and Sugar Alcohols in Energy Drink by Prominence-i with Differential Refractive Index Detector No.L481 Sugars and sugar alcohols display almost no ultraviolet absorption, and are therefore typically detected using a differential refractive index detector or evaporative light uRI scattering detector. By using a ligand exchange column for 80 ■ Peaks sugar analysis, it is possible to distinguish among the different 1. maltose 70 2. glucose isomers based on the position of the hydroxyl group in the 1 4 3. fructose chair conformation of glucose and fructose for example. In 4. erythritol 60 5 other words, the hydroxyl group of the sugar and the metal 5. mannitol ion of the stationary phase form a complex, making it 2 3 6. sorbitol possible to achieve separation due to the difference in the 50 6 strength of the complex formation. Also, maintaining a 40 column temperature of 80 °C suppresses sugar anomer separation and peak dispersion, thereby achieving good 30 separation of adjacent peaks. The new Prominence-i integrated high-performance liquid 20 chromatograph can be connected to the RID-20A differential refractive index detector. The column oven, which can 10 accommodate a 30 cm column and maintain temperature 0 control up to 85 °C, therefore supports applications that require a long column. In Application News No. 467, we introduced an example of 0 5 10 15 20 25 min analysis of sugars in juice, in which the Prominence-i was connected to a differential refractive index detector. Here, we introduce an example of simultaneous analysis of sugars and Fig. -

Novel Insights Into Mannitol Metabolism in the Fungal Plant
Novel insights into mannitol metabolism in the fungal plant pathogen Botrytis cinerea Thierry Dulermo, Christine Rascle, Geneviève Billon-Grand, Elisabeth Gout, Richard Bligny, Pascale Cotton To cite this version: Thierry Dulermo, Christine Rascle, Geneviève Billon-Grand, Elisabeth Gout, Richard Bligny, et al.. Novel insights into mannitol metabolism in the fungal plant pathogen Botrytis cinerea. Biochemical Journal, Portland Press, 2010, 427 (2), pp.323-332. 10.1042/BJ20091813. hal-00479283 HAL Id: hal-00479283 https://hal.archives-ouvertes.fr/hal-00479283 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 05 Feb 2010 as manuscript BJ20091813 1 NOVEL INSIGHTS INTO MANNITOL METABOLISM IN THE FUNGAL PLANT 2 PATHOGEN BOTRYTIS CINEREA 3 4 Authors : Thierry Dulermo*†, Christine Rascle*, Geneviève Billon-Grand*, Elisabeth Gout‡, 5 Richard Bligny‡ and Pascale Cotton§ 6 7 Address 8 *Génomique Fonctionnelle des Champignons Pathogènes des Plantes, UMR 5240 9 Microbiologie, Adaptation -

Sorbitol Malabsorption 1/2
Internal Medicine and Gastroenterology Clinic Dres. Mares,M.Hanig,Mares, Hanig, Rambow, S.Blau, Blau, M.Seip, Hanig, Seip, A.Borchers,Blau,Kirchner, Hochstr. Hochstr. Hochstr. 43, 60313 43, 43, 60313 60313 Frankfurt/Main, Frankfurt/M., Frankfurt/M., www.gastroenterologie-ffm.de www.gastroenterologie-ffm.de www.gastroenterologie-ffm.de Nutrition hints for sorbitol malabsorption 1/2 Sorbitol intolerance Sorbitol incompatibility 1. What does this mean for nutrition? Avoid sorbitol as a sweetener and foods high in sorbitol. Small amount of sorbitol can often be tolerated (at most 10-10 g per day, sometimes less). In order to prevent general upper GI problems, easily digestible foods that do not cause gas are recommended. 2. Key points about foods – Avoid sorbitol and foods containing sorbitol – easily digestible food that does not cause gas 3. vegetables and fruits low in fibre (see 5) 4. Choice of foods The following foods are high in sorbitol and not suitable: – Sorbitol as sweetener: e.g. Sionon, Flarom, diabetic sweetener – Dietetic foods produced with sorbitol: for example, diabetic marmalades, diabetic sweets, diabetic baked goods – Types of fruit which have a naturally high sorbitol content: Apples, pears, cherries, prunes, plums, dates fruits with seeds, such as mirabelles, apricots, nectarines, and all fruit syrups made from these types of fruits – Types of fruit which have a naturally low sorbitol content: Berry fruits such as strawberries, raspberries, blackberries, blueberries, currants, gooseberries, citrus fruits, bananas, pineapples, kiwis – Sorbitol as a coating for: sultanas, raisins, and dried fruit or candied fruit – Sorbitol in sweets: chewing gum, jelly babies, jelly fruits, candies, chocolate bars, filled wafers, chocolate, etc. -

Review of the Role of Mannitol in the Therapy of Children
18th Expert Committee on the Selection and Use of Essential Medicines (21 to 25 March 2011) Section 16: Diuretics -- Mannitol review (Children) Review of the role of mannitol in the therapy of children Dr Fatemeh Tavakkoli M.D. Pharm. D Candidate, University of Maryland, School of Pharmacy, Baltimore, Maryland, USA Contents Introduction......................................................................................................................................... 3 Background ......................................................................................................................................... 3 Table 1: available guidelines for Mannitol ................................................................................ 3 Public Health Burden.......................................................................................................................... 5 Table 2: Indications and dose regimens for Mannitol in children .............................................. 6 Search methods for identification of safety and efficacy data............................................................ 7 Results................................................................................................................................................. 8 Systematic reviews.............................................................................................................................. 8 Individual studies (categorized by therapeutic use of mannitol): ..................................................... 10 -

Copyrighted Material
Index Abbreviations receptor sites 202, 211 weak 122 amino acids, Table 500–1 muscarinic 413 weak, pH calculation 147–8 nucleic acids 551 nicotinic 413 Acid–base nucleotides, Table 551 as quaternary ammonium salt 202 catalysis, enzymes 516 peptides and proteins 504 Acetylcholinesterase equilibria 121 phosphates and diphosphates 277 enzyme mechanism 519–21 interactions, predicting 155–7 structural, Table 14 hydrolysis of acetylcholine 279 Acidic reagents, Table 157 ACE (angiotensin-converting enzyme) inhibitors 279 Acidity enzyme action 532 Acetyl-CoA carboxylase, in fatty acid acidity constant 122 inhibitors 532 biosynthesis 595 bond energy effects 125 Acetal Acetyl-CoA (acetyl coenzyme A) definition 121 in etoposide 233 as acylating agent 262 electronegativity effects 125 formation 229 carboxylation to malonyl-CoA 595, hybridization effects 128 polysaccharides as polyacetals 232 609 inductive effects 125–7 as protecting group 230 Claisen reaction 381 influence of electronic and structural Acetal and ketal enolate anions 373 features 125–34 cyclic, as protecting groups 481 in Krebs cycle 585 and leaving groups, Table 189 groups in sucrose 231 from β-oxidation of fatty acids 388 pKa values 122–5 Acetaldehyde, basicity 139 as thioester 262, 373 resonance / delocalization effects 129–34 Acetamide, basicity 139 Acetylene, bonding molecular orbitals 31 Acidity (compounds) Acetaminophen, see paracetamol Acetylenes, acidity 128 acetone 130 Acetoacetyl-CoA, biosynthesis from N-Acetylgalactosamine, in blood group acetonitrile 365 acetyl-CoA 392 -

United States Patent Office
United States Patentaw Office ... ber of ether groupings on each molecule. These mix tures, however, if they analyze as containing an average 2,978,421. number of ether groups per molecule greater than one, NITRILE COPOLYMERS AND METHOD OF are capable of producing the insoluble nitrile polymers PREPARING SAME of this invention. Since the efficiency of the polyether cross-linking agent increases with the number of poten John A. Holloway, Cleveland, Ohio, assignor to The tially polymerizable groups on the molecule, it is much B. F. Goodrich Company, New York, N.Y., a corpo preferred to utilize polyethers containing an average of ration of New York - two or more alkenyl ether groupings per molecule. The No Drawing. Filed June 9, 1958, Ser. No. 740,566. 10 polyvinyl polyethers of the polyhydric alcohols within the above broad class are produced by reacting acetylene 9 Claims. (C. 260-17.4) with the polyhydric alcohol (or an alcoholate thereof) in a Reppe-type vinylation synthesis. The polycrotyl ethers of the polyhydric alcohols are also useful although they This invention relates to cross-linked alpha-beta ole 15 do not contain a terminal CHCC group. - finically unsaturated nitrile polymers and more particu Illustrative polyhydric alcohols of the above-described larly pertains to copolymers of alpha-beta olefinically class that may be utilized in the preparation of the poly. unsaturated nitriles and polyalkenyl polyethers of poly alkenyl polyether, cross-linking agent include the butane hydric alcohols and methods for their preparation. triols such as 1,2,3-butane triol, 2,3,4-trihydroxybutyric An object of this invention is the provision of insoluble, 20 acid, the aldotetroses such as erythrose and threose, keto cross-linked polynitriles which are capable of thickening tetroses such as erythrulose; the aldopentoses such as certain non-aqueous polar solvents.