Synthesis of Oxa-Bridged Derivatives from Diels–Alder Bis-Adducts of Butadiene and 1,2,3,4-Tetrahalo-5,5-Dimethoxycyclopentadiene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fullerene-Acene Chemistry
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Spring 2007 Fullerene-acene chemistry: Part I Studies on the regioselective reduction of acenes and acene quinones; Part II Progress toward the synthesis of large acenes and their Diels-Alder chemistry with [60]fullerene Andreas John Athans University of New Hampshire, Durham Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation Athans, Andreas John, "Fullerene-acene chemistry: Part I Studies on the regioselective reduction of acenes and acene quinones; Part II Progress toward the synthesis of large acenes and their Diels-Alder chemistry with [60]fullerene" (2007). Doctoral Dissertations. 363. https://scholars.unh.edu/dissertation/363 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. FULLERENE-ACENE CHEMISTRY: PART I: STUDIES ON THE REGIOSELECTIVE REDUCTION OF ACENES AND ACENE QUINONES; PART II: PROGRESS TOWARD THE SYNTHESIS OF LARGE ACENES AND THEIR DIELS- ALDER CHEMISTRY WITH [60]FULLERENE VOLUME 1 CHAPTERS 1-5 BY ANDREAS JOHN ATHANS B.S. University of New Hampshire, 2001 DISSERTATION Submitted to the University of New Hampshire in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy m Chemistry May, 2007 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. UMI Number: 3 2 6 0 5 8 6 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. -

Sulfolane: Magic Extractor Or Bad Actor? Pilot-Scale Study on Solvent Corrosion Potential
sustainability Review Sulfolane: Magic Extractor or Bad Actor? Pilot-Scale Study on Solvent Corrosion Potential Andrzej Bak 1,* , Violetta Kozik 1, Paulina Dybal 1, Slawomir Kus 2, Aleksandra Swietlicka 1 and Josef Jampilek 3,* 1 Department of Synthesis Chemistry, Faculty of Mathematics, Physics and Chemistry, University of Silesia, Szkolna 9, 40007 Katowice, Poland; [email protected] (V.K.); [email protected] (P.D.); [email protected] (A.S.) 2 Honeywell Process Solutions, 11201 Greens Crossing Blvd, Suite 700 Houston, TX 77067, USA; [email protected] 3 Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojarov 10, 83232 Bratislava, Slovakia * Correspondence: [email protected] (A.B.); [email protected] (J.J.); Tel.: +48-32-359-1197 (A.B.) Received: 17 September 2018; Accepted: 11 October 2018; Published: 14 October 2018 Abstract: The sulfur-containing derivatives and their metabolites, regarded as ‘old devils of green’ chemistry, constitute a relevant class of air/water/soil contaminants in over-polluted world. In fact, some industrially-engineered solvents have become environmentally unfavorable. An attractive alternative to commonly used industrial liquids is sulfolane (C4H8SO2), an anthropogenic medium. The main objective of this paper is the comprehensive review focusing mainly on the state-of-the-art aspects of the sulfolane synthesis, application of sulfolane as an extractive solvent due to its ‘unique’ physicochemical properties as well as the potential of sulfolane to cause equipment corrosion and subsequent spills. The potential risk for groundwater contamination, danger for human health and ways of sulfolane biodegradation were briefly reviewed as well. Interestingly, the analysis performed on data stored in the Reaxys database revealed an alternating tendency of waxing and waning interest in sulfolane during the space of the last fifty years. -
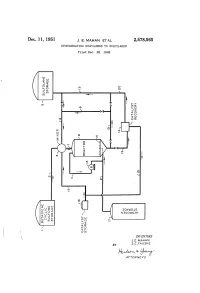
Rudo-At-Thur a 77OAPAVA 1S Patented Dec
Dec. 11, 1951 J. E. MAHAN ET AL 2,578,565 HYDROGENATING SULFOLENES TO SULFOLANES Filed Dec. 28, 1948 o g X g t 92.9 DVOS a dig NGOOOH Ofse INVENTORS J.E. MAHAN BY S.C. FAUSKE Rudo-at-thur A 77OAPAVA 1S Patented Dec. 11, 1951 2,578,565 UNITED STATES PATENT OFFICE 2,578,565 HYDROGENATING SULFOLENESTO SULFOLANES John E. Mahan and Sig C. Fauske, Bartlesville, Okla., assignors to Phillips Petroleum Company, a corporation of Delaware Application December 28, 1948, Serial No. 67,745 15 Claims. (CI. 260-332.1) 1. 2 This invention relates to the production of the generic term “a sulfolane' or "a Sulfolane Sulfolanes by the hydrogenation of the corre compound' covering not only the above Com sponding unsaturated Sulfolenes. In One par pound but also the substituted derivatives there ticular embodiment it relates to an improved of, particularly those in which various radicals process for the production of 2,3,4,5-tetrahy mentioned in the preceding paragraph are Sub drothiophene-1,1-dioxide, commercially known stituted for one or more of the hydrogen atoms as sulfolane, by the catalytic hydrogenation of of the above structure. Where such a radical the corresponding unsaturated cyclic butadiene is hydrogenatable under the conditions of our monosulfone, i. e. 2,5-dihydrothiophene-1,1- process, it will be understood that the sulfolane dioxide, commercially known as Sulfolene, in the 0. containing the hydrogenated radical is included presence of a novel solvent. when reference is made to a sulfolane compound The term “a sulfolene compound' as employed which “corresponds' to a given sulfolene com herein and in the appended claims, defines ge pound. -
Photochemical Reactions As Key Steps in Natural Product Synthesis Thorsten Bach* and J�Rg P
Reviews T. Bach and J. P. Hehn DOI: 10.1002/anie.201002845 Synthetic Methods Photochemical Reactions as Key Steps in Natural Product Synthesis Thorsten Bach* and Jrg P. Hehn Keywords: Dedicated to Professor David A. Evans cyclization · cycloaddition · on the occasion of his 70th birthday natural products · photochemistry · total synthesis Angewandte Chemie 1000 www.angewandte.org 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2011, 50, 1000 – 1045 Photochemical Reactions Photochemical reactions contribute in a significant way to the existing From the Contents repertoire of carbon–carbon bond-forming reactions by allowing access to exceptional molecular structures that cannot be obtained by 1. Introduction 1001 conventional means. In this Review, the most important photochemical 2. Photocyclizations 1001 transformations that have been employed in natural product synthesis are presented. Selected total syntheses are discussed as examples, with 3. Norrish–Yang Cyclizations 1008 particular attention given to the photochemical key step and its ste- reoselectivity. The structural relationship between the photochemically 4. Norrish Type I Cleavage Reactions 1009 generated molecule and the natural product is shown, and, where necessary, the consecutive reactions in the synthesis are illustrated and 5. Photochemical classified. Rearrangements 1011 6. Reactions via Dienol Intermediates 1015 1. Introduction 7. Patern–Bchi Reaction 1017 Is there anything that hasnt already been said or written about natural product synthesis?[1] Great art has been seen in 8. [2+2] Photocycloadditions of it,[2] and attempts have been made to establish it as a Olefins 1018 handcraft. Economical rules have been assigned to it,[3] and it has been fitted into logical schemes.[4] Some people view 9. -

Safety Data Sheet
SAFETY DATA SHEET Creation Date 15-Dec-2011 Revision Date 10-Dec-2020 Revision Number 6 SECTION 1: IDENTIFICATION OF THE SUBSTANCE/MIXTURE AND OF THE COMPANY/UNDERTAKING 1.1. Product identifier Product Description: Butadiene sulfone Cat No. : 107610000; 107610025; 107610050; 107611000; 107615000 Synonyms 3-Sulfolene; 2,5-Dihydrothiophene-1,1-dioxide CAS-No 77-79-2 EC-No. 201-059-7 Molecular Formula C4 H6 O2 S 1.2. Relevant identified uses of the substance or mixture and uses advised against Recommended Use Laboratory chemicals. Uses advised against No Information available 1.3. Details of the supplier of the safety data sheet Company UK entity/business name Fisher Scientific UK Bishop Meadow Road, Loughborough, Leicestershire LE11 5RG, United Kingdom EU entity/business name Acros Organics BVBA Janssen Pharmaceuticalaan 3a 2440 Geel, Belgium E-mail address [email protected] 1.4. Emergency telephone number For information US call: 001-800-ACROS-01 / Europe call: +32 14 57 52 11 Emergency Number US:001-201-796-7100 / Europe: +32 14 57 52 99 CHEMTREC Tel. No.US:001-800-424-9300 / Europe:001-703-527-3887 SECTION 2: HAZARDS IDENTIFICATION 2.1. Classification of the substance or mixture CLP Classification - Regulation (EC) No 1272/2008 Physical hazards Based on available data, the classification criteria are not met Health hazards ______________________________________________________________________________________________ ACR10761 Page 1 / 10 SAFETY DATA SHEET Butadiene sulfone Revision Date 10-Dec-2020 ______________________________________________________________________________________________ Serious Eye Damage/Eye Irritation Category 1 (H318) Environmental hazards Based on available data, the classification criteria are not met Full text of Hazard Statements: see section 16 2.2. -

Grafting of Adipic Anhydride to Carbon Nanotubes Through a Diels-Alder Cycloaddition/Oxidation Cascade Reaction
Grafting of adipic anhydride to carbon nanotubes through a Diels-Alder cycloaddition/oxidation cascade reaction Rui Filipe Araújo a, Maria Fernanda Proença *,b, Carlos Jorge Silva b, Tarsila G. Castro b,c, Manuel Melle-Franco c, Maria Conceição Paiva a, Silvia Vilar-Rodil d, Juan Manuel D. Tascón d a Instituto de Polímeros e Compósitos/I3N, Universidade do Minho, 4800-058 Guimarães, Portugal b Centro de Química, Universidade do Minho, 4710-057 Braga, Portugal c Departamento de Informática, Centro ALGORITMI, Universidade do Minho, 4710-057 Braga, Portugal d Instituto Nacional del Carbón, INCAR-CSIC, Apartado 73, 33080 Oviedo, Spain * Corresponding Author. E-mail address: [email protected] 1 Abstract: Different reactions have been reported for the successful functionalization of carbon nanotubes (CNT). The Diels-Alder cycloaddition is recognized as a plausible chemical approach, but few reports are known where this strategy has been used. In this study, the functionalization was performed by 1,3-butadiene generated from 3-sulfolene under heating conditions in diglyme. This simple and easily scalable method resulted in functionalized CNT with mass losses of 10 - 23 % by thermogravimetric analysis (nitrogen atmosphere). The functionalization was also supported by acid-base titration, elemental analysis, temperature programmed desorption and X-ray photoelectron spectroscopy. The high content in oxygen detected on the CNT surface was assigned to anhydride formation due to a cascade oxidation of the alkene groups generated in the cycloaddition reaction. The complete evolution of the alkene leads to a grafting density of 4.2 mmol g-1 for the anhydride moiety. Ab-initio calculations in CNT model systems indicate that the Diels-Alder addition of butadiene is a feasible process and that subsequent oxidation reactions may result in the formation of the anhydride moiety. -

Sulfolane Technical Assistance and Evaluation Report (PDF)
SULFOLANE TECHNICAL ASSISTANCE AND EVALUATION REPORT FINAL June 1, 2010 Prepared for: Alaska Department of Environmental Conservation Prepared by: 825 W. 8th Ave. Anchorage, AK 99501 Prepared by: Olga Stewart Project Engineer Reviewed by: Lisa Minnear Project Manager - Page Intentionally Left Blank - Sulfolane Technical Assistance and Evaluation Final Report Alaska Department of Environmental Conservation TABLE OF CONTENTS ACRONYMS AND ABBREVIATIONS .............................................................................. v EXECUTIVE SUMMARY ................................................................................................... 1 1. INTRODUCTION .......................................................................................................... 3 2. SULFOLANE PROPERTIES ....................................................................................... 5 2.1. Physical Properties ............................................................................................. 5 2.2. Chemical Properties ........................................................................................... 5 2.3. Water Interaction ................................................................................................ 6 2.4. Breakdown Processes ........................................................................................ 6 2.5. Attenuation Information....................................................................................... 9 3. INDUSTRY USES AND STANDARDS .................................................................... -

ENT OFFICE 2,439,845 YOROCARBON-SUBSTITUTED THIACY - CLOPENTANE-1,1-Dioxide Rupert C
Patented Apr. 6, 1948 2439,345 UNITED STATE S PAT ENT OFFICE 2,439,845 YOROCARBON-SUBSTITUTED THIACY - CLOPENTANE-1,1-DioxIDE Rupert C. Morris, Berkeley, Calif., assignor to Shell Development Company, San Francisco, Calif., a corporation of Delaware No Drawing. Application July 13, 1943 . Serial No. 494,587 - 4 Claims. (C. 260-329) 1 2 This invention relates to a novel and particu as well as the various derivatives thereof, i.e. larly useful class of compounds. More particu sulfolenes in which various radicals are substi larly, the invention pertains to a certain novel tuted for one or more of the hydrogen atoms of and useful class of substituted cyclic sulfones, the above structures. Specifically, the invention is directed to sul It is an object of the present invention to pro folanes and sulfolenes having at least one sub vide a new class of chemical compounds. A fur stituted or unsubstituted unsaturated hydrocar ther object is to provide a novel class of com bon radical attached to a nuclear carbon atom pounds possessing unexpectedly useful proper Of the Sulfolane or sulfolene ring by means of a ties. Other objects will be apparent from the single carbon-to-carbon linkage. O following description of the present invention. The term 'sulfolene,' as employed herein and Unsubstituted sulfoliane, as well as both unsub in the appended claims, refers to an unsaturated stituted sulfolenes, i. e. 2-sulfolene and 3-sul structure containing four carbon atoms, a single folene, have been known for SOme time. Also, olefin linkage between any two adjoining carbon some substituted solfolanes and Sulfolenes have atoms, and a sulfur atom in a ring, the sulfur is been previously prepared. -

United States Patent Office Patiented Feb
3,077,479 United States Patent Office Patiented Feb. 12, 1963 2 The condensation product of sulfur dioxide with 3,077,479 butadiene is, as has been set forth heretofore, sulfolene. PURE FICATION OF SULFGLENE It is preferred to carry out this reaction so as to obtain 3 Daniel B. Lateia, Sr., and Friedrich G. Hefferica, Berke Sulfolene isomer which is more readily hydrogenated than ey, Calif., assignac's to Sheil Gi Co, nasay, New York, 2-sulfoiene. This material, after treatment to deactivate N.Y., a corporation of Delaware catalyst poisons, is then hydrogenated at about 30° C. No Drawing. Fied May 5, 1961, Ser. No. 107,931 over Raney nickel catalyst with a conversion of about 9 Cains. (C. 260-332.1) 97%. In general, this hydrogenation should be carried This invention relates to the manufacture of sulfolane out within about 12 hours, and preferably within about 6 and homologues thereof, particularly lower alkyl Sul 0. hours, following the purification treatment; substantially folanes of up to about eight carbon atoms. More partic immediate hydrogenation is advantageous. ularly, it relates to an improvement in the reaction of 1,3- Sulfolene can also be produced by reacting sulfur di diolefins with sulfur dioxide to form sulfolenes and the oxide and butadiene at about 95 C. in an isopropanol catalytic hydrogenation of the sulfolenes to sulfolanes. solution. At 1:1 mole ratio of sulfur dioxide to buta The sulfolanes are well known solvents useful in extrac 5 diene the conversion to sulfolene of the butadiene is tive distillations, solvent extractions, and the like, especial about 60-65%. -

Butadiene Sulfone As ‘Volatile’, Recyclable Dipolar, Aprotic Solvent for Conducting Substitution and Cycloaddition Reactions Yong Huang1,3†, Esteban E
Huang et al. Sustain Chem Process (2015) 3:13 DOI 10.1186/s40508-015-0040-7 RESEARCH ARTICLE Open Access Butadiene sulfone as ‘volatile’, recyclable dipolar, aprotic solvent for conducting substitution and cycloaddition reactions Yong Huang1,3†, Esteban E. Ureña‑Benavides1,3†, Afrah J. Boigny1, Zachary S. Campbell1, Fiaz S. Mohammed1,3, Jason S. Fisk4, Bruce Holden4, Charles A. Eckert1,2,3, Pamela Pollet2,3* and Charles L. Liotta1,2,3* Abstract Butadiene sulfone has been employed as a “volatile”, recyclable dipolar, aprotic solvent in the reaction of benzyl halide with metal azides to form benzyl azide (1) and the subsequent reaction of benzyl azide with p-toluenesulfonyl cyanide (3) to produce 1-benzyl-5-(p-toluenesulfonyl)tetrazole (2). Comparisons are made with the solvent DMSO and an analogous sulfolene solvent—piperylene sulfone. In addition, recycling protocols for butadiene sulfone and piperylene sulfone are also presented. Keywords: Butadiene sulfone, Piperylene sulfone, Sulfolenes, Recyclable dipolar aprotic solvent, Tetrazoles, Sustainable, Green, DMSO Background undergoes a smooth reversal at 110 °C while butadiene Dimethylsulfoxide (DMSO) is an outstanding solvent sulfone requires temperatures in the 135–140 °C range. for conducting a wide variety of organic reactions. Its In both cases the gaseous diene and SO2 can be captured specific dipolar, aprotic properties allow for the dissolu- by condensing at low temperatures (−76 °C) and react- tion of a range of organic molecules and ionic species. ing at room temperature to reform the original sulfolene Unfortunately, the isolation of reaction products and solvent. the recyclability of the solvent is often times difficult as Several reports have employed sulfolenes as primary well as economically expensive. -

Synthesis, Characterization, and Reactivity of Cross-Conjugated Frameworks
SYNTHESIS, CHARACTERIZATION, AND REACTIVITY OF CROSS-CONJUGATED FRAMEWORKS by Victoria Leigh Hamilton A thesis submitted to the faculty of The University of North Carolina at Charlotte in partial fulfillment of the requirements for the degree of Master of Science in Chemistry Charlotte 2018 Approved by: ______________________________ Markus Etzkorn ______________________________ Craig Ogle ______________________________ Christopher Bejger ______________________________ Douglas Shafer ii ©2018 Victoria Leigh Hamilton ALL RIGHTS RESERVED iii ABSTRACT VICTORIA LEIGH HAMILTON. Synthesis, Characterization, and Reactivity of Cross-Conjugated Frameworks. (Under the direction of DR. MARKUS ETZKORN) Molecules with cross-conjugated -systems possess unique structures and reactivity due to their distinctive form of conjugation that fundamentally differs from the isomeric through conjugated systems by atom connectivity. Many of the organic compounds that played a vital role in the development of industrial organic chemistry are cross-conjugated systems or were transformed to cross-conjugated organic salts (e.g., dyes such as indigo). Our larger goal to synthesize novel cross-conjugated frameworks with potential for biological or materials applications, depended on a preparative scale synthesis of necessary precursors. We prepared a small library of arene-substituted isoindanone derivatives that necessitated optimization of synthetic routes according to electronic demands of the individual systems. Reproducible routes were actualized to afford multigram -

Spectra Libary Index Ichem/Aldrich ATR Library
S p e c t r a L i b r a r y I n d e x Ichem / Aldrich ATR Library ライブラリー名:ATR スタンダードライブラリー 商品番号:30001-40 株式会社 エス・ティ・ジャパン 〒103-0014 東京都中央区日本橋蛎殻町 1-14-10 Tel: 03-3666-2561 Fax:03-3666-2658 http://www.stjapan.co.jp 1 【販売代理店】(株)テクノサイエンス Tel:043-206-0155 Fax:043-206-0188 https://www.techno-lab-co.jp/ (((3,4-DIHYDROXY-9,10-DIOXO-9,10-DIHYDRO-2-ANTHRYL)METHYL)IMINO)DIACETIC ACID ((1,2-DIETHYLETHYLENE)BIS(P-PHENYLENE))DIACETATE ((1-NAPHTHYL)METHYL)AMINE ((1R)-ENDO)-(+)-3-BROMOCAMPHOR, 98 % ((1S)-(ENDO,ANTI))-(-)-3-BROMOCAMPHOR-8-SULFONIC ACID AMMONIUM SALT, 97 % ((1S)-ENDO)-(-)-3-BROMOCAMPHOR, 98 % ((2,4,6-TRIOXOHEXAHYDRO-5-PYRIMIDINYL)IMINO)DIACETIC ACID ((2-CARBOXYETHYL)IMINO)DIACETIC ACID ((2-CYANOETHYLAMINO)(3-INDOLYL)METHYLENE)MALONONITRILE ((2-HYDROXYETHYLAMINO)(3-INDOLYL)METHYLENE)MALONONITRILE ((2-NITROBENZYL)IMINO)DIACETIC ACID ((2-SULFOETHYL)IMINO)DIACETIC ACID ((3-(1-BROMO-1-METHYLETHYL)-7-OXO-1,3,5-CYCLOHEPTATRIEN-1-YL)OXY)DIFLUOROBORANE ((3-INDOLYL)(METHYLTHIO)METHYLENE)MALONONITRILE ((4-NITROBENZYL)IMINO)DIACETIC ACID ((5-ISOPROPYL-2-METHYLCYCLOHEXYL)SULFONYLMETHYL)BENZENE ((N-(2-CYANOETHYL)-N-METHYLAMINO)(3-INDOLYL)METHYLENE)MALONONITRILE ((P-BROMOBENZOYLMETHYLTHIO)METHYLENE)DIMETHYLAMMONIUM BROMIDE ((R)-(+)-2,2'-BIS(DIPHENYLPHOSPHINO)-1,1'-BINAPHTHYL)PALLADIUM(II) CHLORIDE, 97 % ((S)-(-)-2,2'-BIS(DIPHENYLPHOSPHINO)-1,1'-BINAPHTHYL) (1,5-CYCLOOCTADIENE) RHODIUM(I) PERCHLORATE:TETRAHYDROFURAN COMPLEX, 1:1 (-)-(1A,2B,5A)-2-ISOPROPYL-5-METHYLCYCLOHEXYL (1-NAPHTHYL)CARBAMATE (-)-(1A,2B,5A)-2-ISOPROPYL-5-METHYLCYCLOHEXYL 3,5-DINITROBENZOATE