Oxidation of Sulfolane in Aqueous Systems by Chemical and Photochemical Processes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Provisional Peer-Reviewed Toxicity Values for Sulfolane (Casrn 126-33-0)
EPA/690/R-12/024F l Final 1-30-2012 Provisional Peer-Reviewed Toxicity Values for Sulfolane (CASRN 126-33-0) Superfund Health Risk Technical Support Center National Center for Environmental Assessment Office of Research and Development U.S. Environmental Protection Agency Cincinnati, OH 45268 AUTHORS, CONTRIBUTORS, AND REVIEWERS CHEMICAL MANAGER Dan D. Petersen, PhD, DABT National Center for Environmental Assessment, Cincinnati, OH DRAFT DOCUMENT PREPARED BY ICF International 9300 Lee Highway Fairfax, VA 22031 PRIMARY INTERNAL REVIEWERS Ghazi Dannan, PhD National Center for Environmental Assessment, Washington, DC Q. Jay Zhao, PhD, MPH, DABT National Center for Environmental Assessment, Cincinnati, OH This document was externally peer reviewed under contract to Eastern Research Group, Inc. 110 Hartwell Avenue Lexington, MA 02421-3136 Questions regarding the contents of this document may be directed to the U.S. EPA Office of Research and Development’s National Center for Environmental Assessment, Superfund Health Risk Technical Support Center (513-569-7300). i Sulfolane TABLE OF CONTENTS COMMONLY USED ABBREVIATIONS ................................................................................... iii BACKGROUND .............................................................................................................................1 DISCLAIMERS ...............................................................................................................................1 QUESTIONS REGARDING PPRTVS ...........................................................................................1 -

Groundwater and Soil Oxygen and Nutrients (Soil Tilling, Blowers) (Biogenie, 2006)
RemTech 2016 Brent Lennox, M.Sc., P.Geol., Senior Hydrogeologist Eric Pringle, M.A.Sc., P.Eng., Principal Hydrogeological Engineer Waterline Resources Inc. Used in Sulfinol for sour gas sweetening since 1960s Human health related guidelines Poorly adsorbed to soil High solubility in water Microbial degradation slow in typical groundwater conditions Clear, colourless, no field indicators (visual or olefactory) Microbial degradation rapid in aerobic environments and surface water (CCME, 2006) + 2- C4H8O2S + 6.5O2 4CO2 + 3H2O + 2H +SO4 Nutrients improve degradation times Low pH conditions inhibit degradation Typical degradation times: 2 to 4 days at 28°C and 8 to 12 days at 8°C (Green et al., 1998), average air temperatures during trials ranged from 6.9 to 14.1°C Groundwater and Soil Oxygen and nutrients (soil tilling, blowers) (Biogenie, 2006) Groundwater Activated Sludge Treatment System (WorleyParsons Komex, 2008) Oxidants (e.g., hydrogen peroxide) and/or UV light Mixed success (Barr Engineering, 2013; Gallegos et al., 2013; EBA, 2015) Peroxide and iron catalyst shown to be more effective than peroxide (Gallegos, 2013) No sulphate as by-product Site is an operating gas plant located in southern Alberta Constructed in 1960s Sulfolane investigation and monitoring since 1994 No active facilities Downgradient of active facilities Majority of plant waste stored here before the 1980s Potential materials disposed: alumina catalyst, filters (compressor, sulfinol, salt water, glycol, solvent receiver), zeolite, etc. Cells -

Fullerene-Acene Chemistry
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Spring 2007 Fullerene-acene chemistry: Part I Studies on the regioselective reduction of acenes and acene quinones; Part II Progress toward the synthesis of large acenes and their Diels-Alder chemistry with [60]fullerene Andreas John Athans University of New Hampshire, Durham Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation Athans, Andreas John, "Fullerene-acene chemistry: Part I Studies on the regioselective reduction of acenes and acene quinones; Part II Progress toward the synthesis of large acenes and their Diels-Alder chemistry with [60]fullerene" (2007). Doctoral Dissertations. 363. https://scholars.unh.edu/dissertation/363 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. FULLERENE-ACENE CHEMISTRY: PART I: STUDIES ON THE REGIOSELECTIVE REDUCTION OF ACENES AND ACENE QUINONES; PART II: PROGRESS TOWARD THE SYNTHESIS OF LARGE ACENES AND THEIR DIELS- ALDER CHEMISTRY WITH [60]FULLERENE VOLUME 1 CHAPTERS 1-5 BY ANDREAS JOHN ATHANS B.S. University of New Hampshire, 2001 DISSERTATION Submitted to the University of New Hampshire in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy m Chemistry May, 2007 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. UMI Number: 3 2 6 0 5 8 6 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. -
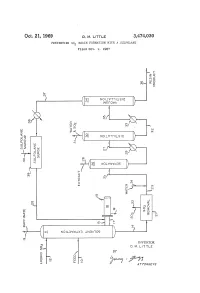
G ( NOW T LSO )
Oct. 21, 1969 D. M. E. 3,474,030 PREVENING SO RESIN FORMATION WITH A SULFOLANE Filed Oct. 9, 1967 f O O n S. NO WISO r W?? WA Q i? 1 a O r Q r C " ? l (? - 2 st e3 r g ( NOW T LSO ) SP & y (T)w 5 > < n Q - ( c O cy P O (? CO w o (f) 7: (s OW&WS ) O) O 3. R k N i 1 ? k y CO s m Yn C - CO s aa ( SS 2 Ele=E c l N 5: N NOLOW X3 NBA OS c INVENTOR. D. M. LTTLE BY y C Year A 77OAPAWAYS 3,474,030 United States Patent Office Patented Oct. 21, 1969 2 A further object of this invention is to minimize the 3,474,030 formation of resinous deposits during the operation of PREVENTING SO, RESIN FORMATION a unit employing SO2 and to provide a practical method WITH A SULFOLANE Donald M. Little, Bartlesville, Okla., assignor to Phillips for removing resinous materials inadvertently formed dur Petroleum Company, a corporation of Delaware 5 ing operation of the process. Filed Oct. 9, 1967, Ser. No. 673,607 Other objects, aspects as well as the several advantages Int. C. C10g 21/10, 21/22 of the invention will be apparent to those skilled in the U.S. C. 208-338 8 Claims art upon reading the specification, drawing, and the ap pended claims. O Summary of the invention ABSTRACT OF THE DISCLOSURE According to the invention, I have found that resinous Dissolution of resinous materials formed from SO2 and materials formed from SO2 and unsaturated materials unsaturated materials coming in contact with SO2, for ex contained in hydrocarbon oils coming in contact with SO ample, during the SO2 solvent extraction of hydrocarbon can be dissolved by contacting with at least one sulfolane. -

Sulfolane: Magic Extractor Or Bad Actor? Pilot-Scale Study on Solvent Corrosion Potential
sustainability Review Sulfolane: Magic Extractor or Bad Actor? Pilot-Scale Study on Solvent Corrosion Potential Andrzej Bak 1,* , Violetta Kozik 1, Paulina Dybal 1, Slawomir Kus 2, Aleksandra Swietlicka 1 and Josef Jampilek 3,* 1 Department of Synthesis Chemistry, Faculty of Mathematics, Physics and Chemistry, University of Silesia, Szkolna 9, 40007 Katowice, Poland; [email protected] (V.K.); [email protected] (P.D.); [email protected] (A.S.) 2 Honeywell Process Solutions, 11201 Greens Crossing Blvd, Suite 700 Houston, TX 77067, USA; [email protected] 3 Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojarov 10, 83232 Bratislava, Slovakia * Correspondence: [email protected] (A.B.); [email protected] (J.J.); Tel.: +48-32-359-1197 (A.B.) Received: 17 September 2018; Accepted: 11 October 2018; Published: 14 October 2018 Abstract: The sulfur-containing derivatives and their metabolites, regarded as ‘old devils of green’ chemistry, constitute a relevant class of air/water/soil contaminants in over-polluted world. In fact, some industrially-engineered solvents have become environmentally unfavorable. An attractive alternative to commonly used industrial liquids is sulfolane (C4H8SO2), an anthropogenic medium. The main objective of this paper is the comprehensive review focusing mainly on the state-of-the-art aspects of the sulfolane synthesis, application of sulfolane as an extractive solvent due to its ‘unique’ physicochemical properties as well as the potential of sulfolane to cause equipment corrosion and subsequent spills. The potential risk for groundwater contamination, danger for human health and ways of sulfolane biodegradation were briefly reviewed as well. Interestingly, the analysis performed on data stored in the Reaxys database revealed an alternating tendency of waxing and waning interest in sulfolane during the space of the last fifty years. -
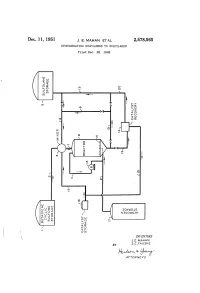
Rudo-At-Thur a 77OAPAVA 1S Patented Dec
Dec. 11, 1951 J. E. MAHAN ET AL 2,578,565 HYDROGENATING SULFOLENES TO SULFOLANES Filed Dec. 28, 1948 o g X g t 92.9 DVOS a dig NGOOOH Ofse INVENTORS J.E. MAHAN BY S.C. FAUSKE Rudo-at-thur A 77OAPAVA 1S Patented Dec. 11, 1951 2,578,565 UNITED STATES PATENT OFFICE 2,578,565 HYDROGENATING SULFOLENESTO SULFOLANES John E. Mahan and Sig C. Fauske, Bartlesville, Okla., assignors to Phillips Petroleum Company, a corporation of Delaware Application December 28, 1948, Serial No. 67,745 15 Claims. (CI. 260-332.1) 1. 2 This invention relates to the production of the generic term “a sulfolane' or "a Sulfolane Sulfolanes by the hydrogenation of the corre compound' covering not only the above Com sponding unsaturated Sulfolenes. In One par pound but also the substituted derivatives there ticular embodiment it relates to an improved of, particularly those in which various radicals process for the production of 2,3,4,5-tetrahy mentioned in the preceding paragraph are Sub drothiophene-1,1-dioxide, commercially known stituted for one or more of the hydrogen atoms as sulfolane, by the catalytic hydrogenation of of the above structure. Where such a radical the corresponding unsaturated cyclic butadiene is hydrogenatable under the conditions of our monosulfone, i. e. 2,5-dihydrothiophene-1,1- process, it will be understood that the sulfolane dioxide, commercially known as Sulfolene, in the 0. containing the hydrogenated radical is included presence of a novel solvent. when reference is made to a sulfolane compound The term “a sulfolene compound' as employed which “corresponds' to a given sulfolene com herein and in the appended claims, defines ge pound. -

A Field Pilot Study on Treating Groundwater Contaminated with Sulfolane Using UV/H2O2
water Article A Field Pilot Study on Treating Groundwater Contaminated with Sulfolane Using UV/H2O2 Linlong Yu 1, Sobhan Iranmanesh 1, Ian Keir 2 and Gopal Achari 1,* 1 Department of Civil Engineering, University of Calgary, 2500 University Drive NW, Calgary, AB T2N 1N4, Canada; [email protected] (L.Y.); [email protected] (S.I.) 2 Bonavista Energy Corporation (formerly), 525 8 Ave SW Suite 1500, Calgary, AB T2P 1G1, Canada; [email protected] * Correspondence: [email protected]; Tel.: +1-403-220-6599 Received: 3 April 2020; Accepted: 21 April 2020; Published: 23 April 2020 Abstract: Sulfolane is an emerging contaminant in the groundwater and soil nearby gas plants, which has attracted much attention from many researchers and regulatory agencies in the past ten years. In this paper, a field pilot-scale ultraviolet (UV)/hydrogen peroxide (H2O2) system was investigated for treating sulfolane contaminated groundwater. Different groundwater, as well as different operational parameters such as influent sulfolane concentration, H2O2 dosage, and water flow rates, were studied. The results showed that a pilot-scale UV/H2O2 system can successfully treat sulfolane contaminated groundwater in the field, although the presence of iron and other groundwater limited the process efficiency. The lowest electrical energy per order of reduction for 3 1 treating sulfolane in groundwater by using the pilot-scale UV/H2O2 system was 1.4 kWh m− order− . The investigated sulfolane initial concentrations and the water flow rates did not impact the sulfolane degradation. The enhancement of sulfolane degradation in an open reservoir by adding ozone was not observed in this study. -

Licensed Technologies Innovative | Proven | Value-Adding
Licensed Technologies Innovative | Proven | Value-adding lyondellbasell.com Saudi Ethylene and Polyethylene Company (SEPC), Al-Jubail Industrial City, Kingdom of Saudi Arabia LyondellBasell is the world’s third-largest independent chemical company. We have annual revenues of approximately $30.8 billion and more than 14,000 employees worldwide. Our vertically integrated facilities, broad product portfolio, manufacturing flexibility, superior technology base and reputation for operational excellence enable us to deliver exceptional value to our customers across the petrochemical chain – from refining to advanced product applications. Product diversity and vertical integration allow LyondellBasell to capture value at every step of the petrochemical chain. Natural Gas Wellhead Crude Liquids Capturing value along the chain Refining Refining Fuels Olefins Olefins Crackers Olefins Aromatics T echnology Propylene Olefin OxyFuels Polyethylene Polypropylene Polybutene-1 Acetyls Ethylene Oxide Styrene Derivatives Oxide Glycols, Glycol Ethers Glycols 2nd Level PP Catalloy Derivatives Compounding Butanediol Glycol Ethers Refining & OxyFuels Olefins & Polyolefins Olefins & Polyolefins Intermediates & Derivatives Technology Americas Europe, Asia & International 2 A global leader in polyolefins and chemicals technology, production and marketing About LyondellBasell LyondellBasell’s technologies are some of the Global capacity positions most reliable, efficient and cost effective in the With major administrative offices in Houston, world. With over 280 licensed -

Sulfolane Mixtures As Ambient Aprotic Polar Solvents
(19) TZZ __T (11) EP 2 778 158 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 17.09.2014 Bulletin 2014/38 C07D 333/48 (2006.01) (21) Application number: 14159793.0 (22) Date of filing: 14.03.2014 (84) Designated Contracting States: (71) Applicant: Advanced Technology Materials, Inc. AL AT BE BG CH CY CZ DE DK EE ES FI FR GB Danbury, CT 06810 (US) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (72) Inventor: Cooper, Emanuel, I. Designated Extension States: Scarsdale, NY 10583 (US) BA ME (74) Representative: ABG Patentes, S.L. (30) Priority: 14.03.2013 US 201361781820 P Avenida de Burgos, 16D Edificio Euromor 28036 Madrid (ES) (54) Sulfolane mixtures as ambient aprotic polar solvents (57) An improved solvent containing sulfolane and at least one dialkyl sulfone, preferably dimethyl sulfone, wherein the improved solvent is a liquid at room temperature and can be used for reaction media and electrochemistry. EP 2 778 158 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 778 158 A1 Description FIELD 5 [0001] The present invention relates generally to an improved solvent for reaction media and electrochemistry. DESCRIPTION OF THE RELATED ART [0002] Sulfolane is a well-known industrial solvent used most often for extractive distillation and to purify natural gas, 10 and also as an aprotic reaction medium and for electrochemical applications. Advantageously, sulfolane has a high dielectric constant, a low toxicity and a very low skin penetration rate, is stable, water soluble, chemically compatible with strong acids and oxidants and bases, has a low decomposition rate, and a very high flash point. -

NTP Research Concept: Sulfolane
NTP Research Concept: Sulfolane Project Leader Chad Blystone, PhD, DABT NIEHS/DNTP/Toxicology Branch Background and Rationale Sulfolane (CAS# 126-33-0) was nominated by the State of Alaska’s Department of Environmental Conservation and Department of Health and Social Services, Mayor of the Fairbanks North Star Borough, Alaska, a Senator from the Alaska State Legislature, and the Agency for Toxic Substances and Disease Registry. Sulfolane is a polar solvent primarily used in the refining of natural gas and petroleum. Other uses include fractionalization of wood tars and curing of epoxy resins. Production of sulfolane was between 10 and 50 million pounds in the United States according the US EPA’s 2006 Inventory Update Rule reporting. Recently sulfolane has been detected in well water within the town of North Pole, Alaska. A plume was identified in groundwater, originating from a petroleum refinery, and the highest concentration measured in wells outside the refinery was 269 ppb as of January 2011. Due to the contamination, residents were being provided bottled water for use and additional wells were created outside the plume area. Sulfolane contamination has also been identified within Canada in areas near sites of natural gas or petroleum refining and may be present as a groundwater contaminant near other sites of refining around the county. A few studies have demonstrated the uptake of sulfolane into vegetation at contaminated sites. In an occupational setting, inhalation and dermal contact could be the primary route(s) of exposure. Animal toxicity data for sulfolane are limited. An inhalation study of aerosolized sulfolane investigated potential toxic effects in various species (rats, guinea pigs, dogs, squirrel monkeys), but experimental detail and design are limited (1). -
Photochemical Reactions As Key Steps in Natural Product Synthesis Thorsten Bach* and J�Rg P
Reviews T. Bach and J. P. Hehn DOI: 10.1002/anie.201002845 Synthetic Methods Photochemical Reactions as Key Steps in Natural Product Synthesis Thorsten Bach* and Jrg P. Hehn Keywords: Dedicated to Professor David A. Evans cyclization · cycloaddition · on the occasion of his 70th birthday natural products · photochemistry · total synthesis Angewandte Chemie 1000 www.angewandte.org 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2011, 50, 1000 – 1045 Photochemical Reactions Photochemical reactions contribute in a significant way to the existing From the Contents repertoire of carbon–carbon bond-forming reactions by allowing access to exceptional molecular structures that cannot be obtained by 1. Introduction 1001 conventional means. In this Review, the most important photochemical 2. Photocyclizations 1001 transformations that have been employed in natural product synthesis are presented. Selected total syntheses are discussed as examples, with 3. Norrish–Yang Cyclizations 1008 particular attention given to the photochemical key step and its ste- reoselectivity. The structural relationship between the photochemically 4. Norrish Type I Cleavage Reactions 1009 generated molecule and the natural product is shown, and, where necessary, the consecutive reactions in the synthesis are illustrated and 5. Photochemical classified. Rearrangements 1011 6. Reactions via Dienol Intermediates 1015 1. Introduction 7. Patern–Bchi Reaction 1017 Is there anything that hasnt already been said or written about natural product synthesis?[1] Great art has been seen in 8. [2+2] Photocycloadditions of it,[2] and attempts have been made to establish it as a Olefins 1018 handcraft. Economical rules have been assigned to it,[3] and it has been fitted into logical schemes.[4] Some people view 9. -

Safety Data Sheet
SAFETY DATA SHEET Creation Date 15-Dec-2011 Revision Date 10-Dec-2020 Revision Number 6 SECTION 1: IDENTIFICATION OF THE SUBSTANCE/MIXTURE AND OF THE COMPANY/UNDERTAKING 1.1. Product identifier Product Description: Butadiene sulfone Cat No. : 107610000; 107610025; 107610050; 107611000; 107615000 Synonyms 3-Sulfolene; 2,5-Dihydrothiophene-1,1-dioxide CAS-No 77-79-2 EC-No. 201-059-7 Molecular Formula C4 H6 O2 S 1.2. Relevant identified uses of the substance or mixture and uses advised against Recommended Use Laboratory chemicals. Uses advised against No Information available 1.3. Details of the supplier of the safety data sheet Company UK entity/business name Fisher Scientific UK Bishop Meadow Road, Loughborough, Leicestershire LE11 5RG, United Kingdom EU entity/business name Acros Organics BVBA Janssen Pharmaceuticalaan 3a 2440 Geel, Belgium E-mail address [email protected] 1.4. Emergency telephone number For information US call: 001-800-ACROS-01 / Europe call: +32 14 57 52 11 Emergency Number US:001-201-796-7100 / Europe: +32 14 57 52 99 CHEMTREC Tel. No.US:001-800-424-9300 / Europe:001-703-527-3887 SECTION 2: HAZARDS IDENTIFICATION 2.1. Classification of the substance or mixture CLP Classification - Regulation (EC) No 1272/2008 Physical hazards Based on available data, the classification criteria are not met Health hazards ______________________________________________________________________________________________ ACR10761 Page 1 / 10 SAFETY DATA SHEET Butadiene sulfone Revision Date 10-Dec-2020 ______________________________________________________________________________________________ Serious Eye Damage/Eye Irritation Category 1 (H318) Environmental hazards Based on available data, the classification criteria are not met Full text of Hazard Statements: see section 16 2.2.