ARTICLE Submicroscopic Deletion in Patients with Williams-Beuren Syndrome Influences Expression Levels of the Nonhemizygous Flanking Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Predicting Gene Ontology Biological Process from Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R
Methods Predicting Gene Ontology Biological Process From Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R. Hvidsten,2 Herman Midelfart,2 Jan Komorowski,2,3,4 and Arne K. Sandvik1 1Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, N-7489 Trondheim, Norway; 2Department of Information and Computer Science, Norwegian University of Science and Technology, N-7491 Trondheim, Norway; 3The Linnaeus Centre for Bioinformatics, Uppsala University, SE-751 24 Uppsala, Sweden The aim of the present study was to generate hypotheses on the involvement of uncharacterized genes in biological processes. To this end,supervised learning was used to analyz e microarray-derived time-series gene expression data. Our method was objectively evaluated on known genes using cross-validation and provided high-precision Gene Ontology biological process classifications for 211 of the 213 uncharacterized genes in the data set used. In addition,new roles in biological process were hypothesi zed for known genes. Our method uses biological knowledge expressed by Gene Ontology and generates a rule model associating this knowledge with minimal characteristic features of temporal gene expression profiles. This model allows learning and classification of multiple biological process roles for each gene and can predict participation of genes in a biological process even though the genes of this class exhibit a wide variety of gene expression profiles including inverse coregulation. A considerable number of the hypothesized new roles for known genes were confirmed by literature search. In addition,many biological process roles hypothesi zed for uncharacterized genes were found to agree with assumptions based on homology information. -
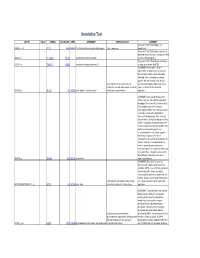
Supplementary Table 5. Functional Annotation of the Largest Gene Cluster(221 Element)
Annotation Tool AFFYID VALUE SYMBOL LOCUSLINK OMIM GENENAME GENEONTOLOGY SUMMARY [Proteome FUNCTION:] Expressed 203054_s_at TCTA 6988 600690 T-cell leukemia translocation altered gene tumor suppressor ubiquitously [Proteome FUNCTION:] May be involved in protein-protein interactions; contains five WD 44563_at FLJ10385 55135 hypothetical protein FLJ10385 domains (WD-40 repeats) [Proteome FUNCTION:] Weakly similarity to 212261_at TNRC15 26058 trinucleotide repeat containing 15 a region of rat nestin (Rn.9701) [SUMMARY:] Actin alpha 1 which is expressed in skeletal muscle is one of six different actin isoforms which have been identified. Actins are highly conserved proteins that are involved in cell motility, actin filament; motor activity; muscle structure and integrity. Alpha actins are a contraction; muscle development; structural major constituent of the contractile 203872_at ACTA1 58 102610 actin, alpha 1, skeletal muscle constituent of cytoskeleton apparatus. [SUMMARY:] Annexin VIII belong to the family of Ca (2+) dependent phospholipid binding proteins (annexins), and has a high 56% identity to annexin V (vascular anticoagulant-alpha). It was initially isolated as 2.2 kb vascular anticoagulant-beta transcript from human placenta, a Ca (2+) dependent phospholipid binding protein that inhibits coagulation and phospholipase A2 activity. However, the fact that annexin VIII is neither an extracellular protein nor associated with the cell surface suggests that it may not play a role in blood coagulation in vivo and its physiological role remains unknown. It is expressed at low levels in human placenta and shows restricted expression in lung endothelia, skin, liver, and kidney. The gene is also found to be selectively overexpressed in acute 203074_at ANXA8 244 602396 annexin A8 myelocytic leukemia. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0297536A1 Chin Et Al
US 20090297536A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0297536A1 Chin et al. (43) Pub. Date: Dec. 3, 2009 (54) COMPOSITIONS, KITS, AND METHODS FOR Related U.S. Application Data IDENTIFICATION, ASSESSMENT, (60) Provisional application No. 60/575,795, filed on May PREVENTION AND THERAPY OF CANCER 28, 2004, provisional application No. 60/580,337, filed on Jun. 15, 2004. (75) Inventors: Lynda Chin, Brookline, MA (US); Publication Classification Cameron W. Brennan, New York, NY (US); Ronald A. DePinho, (51) Int. Cl. Brookline, MA (US); Andrew J. A 6LX 39/395 (2006.01) Aguirre, Boston, MA (US) CI2O I/68 (2006.01) C40B 30/00 (2006.01) AOIK 67/00 (2006.01) Correspondence Address: A 6LX 3L/7052 (2006.01) FOLEY HOAG, LLP A638/02 (2006.01) PATENT GROUP, WORLD TRADE CENTER A638/16 (2006.01) WEST C07K I4/00 (2006.01) 155 SEAPORT BLVD C7H 2L/00 (2006.01) BOSTON, MA 02110 (US) C07K 6/00 (2006.01) A6IP35/00 (2006.01) (73) Assignee: Dana-Farber Cancer Institute, (52) U.S. Cl. ................ 424/172.1; 435/6:506/7; 800/10: Inc., Boston, MA (US) 424/183.1; 424/178.1: 514/44 A: 514/2: 514/12: 530/350:536/23.1; 530/389.1 (21) Appl. No.: 11/597,825 (57) ABSTRACT The invention relates to compositions, kits, and methods for (22) PCT Filed: May 27, 2005 detecting, characterizing, preventing, and treating human cancer. A variety of chromosomal regions (MCRs) and mark (86). PCT No.: PCT/US05/18850 ers corresponding thereto, are provided, wherein alterations in the copy number of one or more of the MCRs and/or S371 (c)(1), alterations in the amount, structure, and/or activity of one or (2), (4) Date: Jun. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Regulation of COX Assembly and Function by Twin CX9C Proteins—Implications for Human Disease
cells Review Regulation of COX Assembly and Function by Twin CX9C Proteins—Implications for Human Disease Stephanie Gladyck 1, Siddhesh Aras 1,2, Maik Hüttemann 1 and Lawrence I. Grossman 1,2,* 1 Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, Detroit, MI 48201, USA; [email protected] (S.G.); [email protected] (S.A.); [email protected] (M.H.) 2 Perinatology Research Branch, Division of Obstetrics and Maternal-Fetal Medicine, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, U.S. Department of Health and Human Services, Bethesda, Maryland and Detroit, MI 48201, USA * Correspondence: [email protected] Abstract: Oxidative phosphorylation is a tightly regulated process in mammals that takes place in and across the inner mitochondrial membrane and consists of the electron transport chain and ATP synthase. Complex IV, or cytochrome c oxidase (COX), is the terminal enzyme of the electron transport chain, responsible for accepting electrons from cytochrome c, pumping protons to contribute to the gradient utilized by ATP synthase to produce ATP, and reducing oxygen to water. As such, COX is tightly regulated through numerous mechanisms including protein–protein interactions. The twin CX9C family of proteins has recently been shown to be involved in COX regulation by assisting with complex assembly, biogenesis, and activity. The twin CX9C motif allows for the import of these proteins into the intermembrane space of the mitochondria using the redox import machinery of Mia40/CHCHD4. Studies have shown that knockdown of the proteins discussed in this review results in decreased or completely deficient aerobic respiration in experimental models ranging from yeast to human cells, as the proteins are conserved across species. -

Investigating Host Genes Involved In. HIY Control by a Novel Computational Method to Combine GWAS with Eqtl
Investigating Host Genes Involved in. HIY Control by a Novel Computational Method to Combine GWAS with eQTL by Yi Song THESIS Submitted In partial satisfaction of me teqoitements for the degree of MASTER OF SCIENCE In Biological and Medical Informatics In the GRADUATE DIVISION Copyright (2012) by Yi Song ii Acknowledgement First and foremost, I would like to thank my advisor Professor Hao Li, without whom this thesis would not have been possible. I am very grateful that Professor Li lead me into the field of human genomics and gave me the opportunity to pursue this interesting study in his laboratory. Besides the wealth of knowledge and invaluable insights that he offered in every meeting we had, Professor Li is one of the most approachable faculties I have met. I truly appreciate his patient guidance and his enthusiastic supervision throughout my master’s career. I am sincerely thankful to Professor Patricia Babbitt, the Associate Director of the Biomedical Informatics program at UCSF. Over my two years at UCSF, she has always been there to offer her help when I was faced with difficulties. I would also like to thank both Professor Babbitt and Professor Nevan Krogan for investing their valuable time in evaluating my work. I take immense pleasure in thanking my co-workers Dr. Xin He and Christopher Fuller. It has been a true enjoyment to discuss science with Dr. He, whose enthusiasm is a great inspiration to me. I also appreciate his careful editing of my thesis. Christopher Fuller, a PhD candidate in the Biomedical Informatics program, has provided great help for me on technical problems. -

(12) Patent Application Publication (10) Pub. No.: US 2009/00992.59 A1 Jouni Et Al
US 200900992.59A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/00992.59 A1 Jouni et al. (43) Pub. Date: Apr. 16, 2009 (54) METHOD FOR REGULATING GENE Related U.S. Application Data EXPRESSION (60) Provisional application No. 60/777,724, filed on Feb. (76) Inventors: Zeina Jouni, Evansville, IN (US); 28, 2006. Joshua Anthony, Evansville, IN (US); Steven C. Rumsey, Curitiba Publication Classification (BR): Deshanie Rai, Evansville, IN (US); Kumar Sesha Kothapalli, (51) Int. Cl. Ithaca, NY (US); James T. Brenna, A6II 3L/20 (2006.01) Ithaca, NY (US) A6IP3/00 (2006.01) A6IP 25/00 (2006.01) Correspondence Address: A6IP37/00 (2006.01) Richard D. Schmidt Bristol-Myers Squibb Company (52) U.S. Cl. ........................................................ 514/560 2400 West Lloyd Expressay Evansville, IN 47721-0001 (US) (57) ABSTRACT (21) Appl. No.: 11/712,102 The present invention is directed to a novel method for modu lating the expression of one or more genes in a subject by (22) Filed: Feb. 28, 2007 administering an amount of DHA and ARA to the subject. ::::::: Patent Application Publication Apr. 16, 2009 US 2009/00992.59 A1 s :::::::::: US 2009/00992.59 A1 Apr. 16, 2009 METHOD FOR REGULATING GENE expression of certain genes in Subjects and thereby prevent EXPRESSION the onset of or treat various diseases and disorders. SUMMARY OF THE INVENTION 0001. This application claims the priority benefit of U.S. 0011 Briefly, the present invention is directed to a novel Provisional Application 60/777,724 filed Feb. 28, 2006 which method for modulating the expression of one or more genes in is incorporated by reference herein in its entirety. -

BRAZILIAN SYMPOSIUM on BIOINFORMATICS August, 29 - 31, 2007 Angra Dos Reis – Rio De Janeiro Brasil
BRAZILIAN SYMPOSIUM ON BIOINFORMATICS August, 29 - 31, 2007 Angra dos Reis – Rio de Janeiro Brasil BSB 2007 Poster Proceedings Conference Chair Sérgio Lifschtiz Department of Informatics - Pontificial Catholic University/Rio de Janeiro Brazil Volume Editors Marie-France Sagot – INRIA – France Maria Emília M. T. Walter – University of Brasília – Brazil Promotion Brazilian Computer Society – SBC Brazilian Symposium on Bioinformatics (2007 : Angra dos Reis, RJ) B827 BSB 2007 poster proceedings, August, 29-31, 2007, Angra dos Reis, Rio de Janeiro, Brasil / conference chair Sérgio Lifschtiz ; volume editors Marie-France Sagot, Maria Emília M. T. Walter. -- [Angra dos Reis] : Brazilian Computer Society, [2007]. 1 CD-ROM. ISBN 978-85-7669-123-5 1. Bioinformática – Congresso. I. Lifschtiz , Sérgio. II. Sagot, Marie-France. III. Walter, Maria Emília M. T. IV. Título. CDD21 004 CDD21 570 Preface The Brazilian Symposium on Bioinformatics (BSB 2007) was held in Angra dos Reis (Rio de Janeiro), Brazil, August 29-31, 2007, on the Portogalo Suite Hotel. It is an event promoted by the Brazilian Computer Society’s (SBC) special committee for computational biology (CEBioComp). BSB 2007 was the second BSB symposium, though BSB is a new name for a predecessor event called Brazilian Workshop on Bioinformatics (WOB). This previous event had three consecutive editions in 2002 (Gramado, Rio Grande do Sul), 2003 (Maca´e,Rio de Janeiro), and 2004 (Brasilia, Distrito Federal). The change from workshop to symposium reflects the increased quality and interest of the meeting. BSB 2007 was held co-located with the International Workshop on Genomic Databases (IWGD 2007). For BSB 2007, we had 60 submissions: 36 full papers and 24 extended ab- stracts, submitted to two tracks, computational biology/bioinformatics and ap- plications. -

High-Resolution Genomic Copy Number Profiling of Glioblastoma Multiforme by Single Nucleotide Polymorphism DNA Microarray
Published OnlineFirst May 12, 2009; DOI: 10.1158/1541-7786.MCR-08-0270 Published Online First on May 12, 2009 High-Resolution Genomic Copy Number Profiling of Glioblastoma Multiforme by Single Nucleotide Polymorphism DNA Microarray Dong Yin,1 Seishi Ogawa,3 Norihiko Kawamata,1 Patrizia Tunici,2 Gaetano Finocchiaro,4 Marica Eoli,4 Christian Ruckert,6 Thien Huynh,1 Gentao Liu,2 Motohiro Kato,3 Masashi Sanada,3 Anna Jauch,5 Martin Dugas,6 Keith L. Black,2 and H. Phillip Koeffler1 1Division of Hematology/Oncology and 2Maxine Dunitz Neurosurgical Institute, Cedars-Sinai Medical Center, University of California at Los Angeles School of Medicine, Los Angeles, California; 3Regeneration Medicine of Hematopoiesis, University of Tokyo, School of Medicine, Tokyo, Japan; 4National Neurological Institute “C Besta,” Milan, Italy; 5Institute of Human Genetics, University Hospital Heidelberg, Germany; and 6Department of Medical Informatics and Biomathematics, University of Munster, Munster, Germany Abstract growth factor receptor/platelet-derived growth factor receptor Glioblastoma multiforme (GBM) is an extremely malignant α. Deletion of chromosome 6q26-27 often occurred (16 of 55 brain tumor. To identify new genomic alterations in GBM, samples). The minimum common deleted region included genomic DNA of tumor tissue/explants from 55 individuals PARK2, PACRG, QKI,and PDE10A genes. Further reverse and 6 GBM cell lines were examined using single nucleotide transcription Q-PCR studies showed that PARK2 expression polymorphism DNA microarray (SNP-Chip). Further gene was decreased in another collection of GBMs at a expression analysis relied on an additional 56 GBM samples. frequency of 61% (34 of 56) of samples. The 1p36.23 region SNP-Chip results were validated using several techniques, was deleted in 35% (19 of 55) of samples. -

(ENO1), Identified As an Antigen to Monoclonal Antibody 12C7
Shu et al. Stem Cell Research & Therapy (2021) 12:119 https://doi.org/10.1186/s13287-021-02160-9 RESEARCH Open Access Alpha-enolase (ENO1), identified as an antigen to monoclonal antibody 12C7, promotes the self-renewal and malignant phenotype of lung cancer stem cells by AMPK/mTOR pathway Xiong Shu1†, Kai-Yue Cao2†, Hui-Qi Liu3, Long Yu4, Li-Xin Sun4, Zhi-Hua Yang4, Cheng-Ai Wu1* and Yu-Liang Ran4* Abstract Background: Tumor-associated antigens (TAAs) can be targeted in cancer therapy. We previously identified a monoclonal antibody (mAb) 12C7, which presented anti-tumor activity in lung cancer stem cells (LCSCs). Here, we aimed to identify the target antigen for 12C7 and confirm its role in LCSCs. Methods: Immunofluorescence was used for antigen localization. After targeted antigen purification by electrophoresis and immunoblot, the antigen was identified by LC-MALDI-TOF/TOF mass spectrometry, immunofluorescence, and immunoprecipitation. The overexpression or silence of ENO1 was induced by lentiviral transduction. Self-renewal, growth, and invasion of LCSCs were evaluated by sphere formation, colony formation, and invasion assay, respectively. High-throughput transcriptome sequencing (RNA-seq) and bioinformatics analysis were performed to analyze downstream targets and pathways of targeted antigen. Results: Targeted antigen showed a surface antigen expression pattern, and the 43–55 kDa protein band was identified as α-enolase (ENO1). Self-renewal, growth, and invasion abilities of LCSCs were remarkably inhibited by ENO1 downregulation, while enhanced by ENO1 upregulation. RNA-seq and bioinformatics analysis eventually screened 4 self-renewal-related and 6 invasion-related differentially expressed genes. GSEA analysis and qRT-PCR verified that ENO1 regulated self-renewal, invasion-related genes, and pathways. -

A Network Inference Approach to Understanding Musculoskeletal
A NETWORK INFERENCE APPROACH TO UNDERSTANDING MUSCULOSKELETAL DISORDERS by NIL TURAN A thesis submitted to The University of Birmingham for the degree of Doctor of Philosophy College of Life and Environmental Sciences School of Biosciences The University of Birmingham June 2013 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Musculoskeletal disorders are among the most important health problem affecting the quality of life and contributing to a high burden on healthcare systems worldwide. Understanding the molecular mechanisms underlying these disorders is crucial for the development of efficient treatments. In this thesis, musculoskeletal disorders including muscle wasting, bone loss and cartilage deformation have been studied using systems biology approaches. Muscle wasting occurring as a systemic effect in COPD patients has been investigated with an integrative network inference approach. This work has lead to a model describing the relationship between muscle molecular and physiological response to training and systemic inflammatory mediators. This model has shown for the first time that oxygen dependent changes in the expression of epigenetic modifiers and not chronic inflammation may be causally linked to muscle dysfunction.