Senescence Signaling, Regulation and Bypass by Telomere Maintenance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -
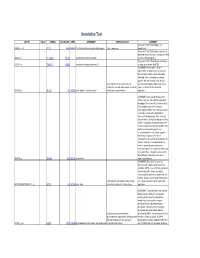
Supplementary Table 5. Functional Annotation of the Largest Gene Cluster(221 Element)
Annotation Tool AFFYID VALUE SYMBOL LOCUSLINK OMIM GENENAME GENEONTOLOGY SUMMARY [Proteome FUNCTION:] Expressed 203054_s_at TCTA 6988 600690 T-cell leukemia translocation altered gene tumor suppressor ubiquitously [Proteome FUNCTION:] May be involved in protein-protein interactions; contains five WD 44563_at FLJ10385 55135 hypothetical protein FLJ10385 domains (WD-40 repeats) [Proteome FUNCTION:] Weakly similarity to 212261_at TNRC15 26058 trinucleotide repeat containing 15 a region of rat nestin (Rn.9701) [SUMMARY:] Actin alpha 1 which is expressed in skeletal muscle is one of six different actin isoforms which have been identified. Actins are highly conserved proteins that are involved in cell motility, actin filament; motor activity; muscle structure and integrity. Alpha actins are a contraction; muscle development; structural major constituent of the contractile 203872_at ACTA1 58 102610 actin, alpha 1, skeletal muscle constituent of cytoskeleton apparatus. [SUMMARY:] Annexin VIII belong to the family of Ca (2+) dependent phospholipid binding proteins (annexins), and has a high 56% identity to annexin V (vascular anticoagulant-alpha). It was initially isolated as 2.2 kb vascular anticoagulant-beta transcript from human placenta, a Ca (2+) dependent phospholipid binding protein that inhibits coagulation and phospholipase A2 activity. However, the fact that annexin VIII is neither an extracellular protein nor associated with the cell surface suggests that it may not play a role in blood coagulation in vivo and its physiological role remains unknown. It is expressed at low levels in human placenta and shows restricted expression in lung endothelia, skin, liver, and kidney. The gene is also found to be selectively overexpressed in acute 203074_at ANXA8 244 602396 annexin A8 myelocytic leukemia. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0297536A1 Chin Et Al
US 20090297536A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0297536A1 Chin et al. (43) Pub. Date: Dec. 3, 2009 (54) COMPOSITIONS, KITS, AND METHODS FOR Related U.S. Application Data IDENTIFICATION, ASSESSMENT, (60) Provisional application No. 60/575,795, filed on May PREVENTION AND THERAPY OF CANCER 28, 2004, provisional application No. 60/580,337, filed on Jun. 15, 2004. (75) Inventors: Lynda Chin, Brookline, MA (US); Publication Classification Cameron W. Brennan, New York, NY (US); Ronald A. DePinho, (51) Int. Cl. Brookline, MA (US); Andrew J. A 6LX 39/395 (2006.01) Aguirre, Boston, MA (US) CI2O I/68 (2006.01) C40B 30/00 (2006.01) AOIK 67/00 (2006.01) Correspondence Address: A 6LX 3L/7052 (2006.01) FOLEY HOAG, LLP A638/02 (2006.01) PATENT GROUP, WORLD TRADE CENTER A638/16 (2006.01) WEST C07K I4/00 (2006.01) 155 SEAPORT BLVD C7H 2L/00 (2006.01) BOSTON, MA 02110 (US) C07K 6/00 (2006.01) A6IP35/00 (2006.01) (73) Assignee: Dana-Farber Cancer Institute, (52) U.S. Cl. ................ 424/172.1; 435/6:506/7; 800/10: Inc., Boston, MA (US) 424/183.1; 424/178.1: 514/44 A: 514/2: 514/12: 530/350:536/23.1; 530/389.1 (21) Appl. No.: 11/597,825 (57) ABSTRACT The invention relates to compositions, kits, and methods for (22) PCT Filed: May 27, 2005 detecting, characterizing, preventing, and treating human cancer. A variety of chromosomal regions (MCRs) and mark (86). PCT No.: PCT/US05/18850 ers corresponding thereto, are provided, wherein alterations in the copy number of one or more of the MCRs and/or S371 (c)(1), alterations in the amount, structure, and/or activity of one or (2), (4) Date: Jun. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Understanding Brassicaceae Evolution Through Ancestral
Understanding Brassicaceae evolution through ancestral genome reconstruction Florent Murat, Alexandra Louis, Florian Maumus, Alix Armero Villanueva, Richard Cooke, Hadi Quesneville, Hugues Roest Crollius, Jerome Salse To cite this version: Florent Murat, Alexandra Louis, Florian Maumus, Alix Armero Villanueva, Richard Cooke, et al.. Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biology, BioMed Central, 2015, 16 (262), 10.1186/s13059-015-0814-y. hal-01281832 HAL Id: hal-01281832 https://hal.archives-ouvertes.fr/hal-01281832 Submitted on 2 Mar 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Murat et al. Genome Biology (2015) 16:262 DOI 10.1186/s13059-015-0814-y RESEARCH Open Access Understanding Brassicaceae evolution through ancestral genome reconstruction Florent Murat1†, Alexandra Louis2,3,4†, Florian Maumus5†, Alix Armero1, Richard Cooke6, Hadi Quesneville5, Hugues Roest Crollius2,3,4 and Jerome Salse1* Abstract Background: Brassicaceae is a family of green plants of high scientific and economic interest, including thale cress (Arabidopsis thaliana), cruciferous vegetables (cabbages) and rapeseed. Results: We reconstruct an evolutionary framework of Brassicaceae composed of high-resolution ancestral karyotypes using the genomes of modern A. thaliana, Arabidopsis lyrata, Capsella rubella, Brassica rapa and Thellungiella parvula. -

1 Human Cells Contain Myriad Excised Linear Intron Rnas with Potential
Human cells contain myriad excised linear intron RNAs with potential functions in gene regulation and as disease biomarkers Jun Yao,1 Hengyi Xu,1 Shelby Winans,1 Douglas C. Wu,1,3 Manuel Ares, Jr.,2 and Alan M. Lambowitz1,4 1Institute for Cellular and Molecular Biology and Departments of Molecular Biosciences and Oncology University of Texas at Austin Austin TX 78712 2Department of Molecular, Cell, and Developmental Biology University of California, Santa Cruz Santa Cruz, California 95064 Running Title: Structured excised linear intron RNAs 3Present address: Invitae Corporation 4To whom correspondence should be addressed. E-mail: [email protected] 1 Abstract We used thermostable group II intron reverse transcriptase sequencing (TGIRT-seq), which gives full-length end-to-end sequence reads of structured RNAs, to identify > 8,500 short full- length excised linear intron (FLEXI) RNAs originating from > 3,500 different genes in human cells and tissues. Most FLEXI RNAs have stable predicted secondary structures, making them difficult to detect by other methods. Some FLEXI RNAs corresponded to annotated mirtron pre- miRNAs (introns that are processed by DICER into functional miRNAs) or agotrons (introns that bind AGO2 and function in a miRNA-like manner) and a few encode snoRNAs. However, the vast majority had not been characterized previously. FLEXI RNA profiles were cell-type specific, reflecting differences in host gene transcription, alternative splicing, and intron RNA turnover, and comparisons of matched tumor and healthy tissues from breast cancer patients and cell lines revealed hundreds of differences in FLEXI RNA expression. About half of the FLEXI RNAs contained an experimentally identified binding site for one or more proteins in published CLIP-seq datasets. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Molecular Diagnostic Requisition
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 MOLECULAR DIAGNOSTIC REQUISITION PATIENT INFORMATION SAMPLE INFORMATION NAME: DATE OF COLLECTION: / / LAST NAME FIRST NAME MI MM DD YY HOSPITAL#: ACCESSION#: DATE OF BIRTH: / / GENDER (Please select one): FEMALE MALE MM DD YY SAMPLE TYPE (Please select one): ETHNIC BACKGROUND (Select all that apply): UNKNOWN BLOOD AFRICAN AMERICAN CORD BLOOD ASIAN SKELETAL MUSCLE ASHKENAZIC JEWISH MUSCLE EUROPEAN CAUCASIAN -OR- DNA (Specify Source): HISPANIC NATIVE AMERICAN INDIAN PLACE PATIENT STICKER HERE OTHER JEWISH OTHER (Specify): OTHER (Please specify): REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: INDICATION FOR STUDY SYMPTOMATIC (Summarize below.): *FAMILIAL MUTATION/VARIANT ANALYSIS: COMPLETE ALL FIELDS BELOW AND ATTACH THE PROBAND'S REPORT. GENE NAME: ASYMPTOMATIC/POSITIVE FAMILY HISTORY: (ATTACH FAMILY HISTORY) MUTATION/UNCLASSIFIED VARIANT: RELATIONSHIP TO PROBAND: THIS INDIVIDUAL IS CURRENTLY: SYMPTOMATIC ASYMPTOMATIC *If family mutation is known, complete the FAMILIAL MUTATION/ VARIANT ANALYSIS section. NAME OF PROBAND: ASYMPTOMATIC/POPULATION SCREENING RELATIONSHIP TO PROBAND: OTHER (Specify clinical findings below): BMGL LAB#: A COPY OF ORIGINAL RESULTS ATTACHED IF PROBAND TESTING WAS PERFORMED AT ANOTHER LAB, CALL TO DISCUSS PRIOR TO SENDING SAMPLE. A POSITIVE CONTROL MAY BE REQUIRED IN SOME CASES. REQUIRED: NEW YORK STATE PHYSICIAN SIGNATURE OF CONSENT I certify that the patient specified above and/or their legal guardian has been informed of the benefits, risks, and limitations of the laboratory test(s) requested. -

Adenylate Kinase 2 / AK2 (1-239, His-Tag) Human Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AR09002PU-N Adenylate kinase 2 / AK2 (1-239, His-tag) Human Protein Product data: Product Type: Recombinant Proteins Description: Adenylate kinase 2 / AK2 (1-239, His-tag) human recombinant protein, 0.5 mg Species: Human Expression Host: E. coli Tag: His-tag Predicted MW: 28.6 kDa Concentration: lot specific Purity: >95% by SDS-PAGE Buffer: Presentation State: Purified State: Liquid purified protein Buffer System: 20 mM Tris pH 7.5, 5 mM DTT, 20% glycerol Bioactivity: Specific: > 1.5 units/ml One unit will convert 2.0 umoles of ADP to ATP + AMP per minute at pH 7.5 at 25°C. Endotoxin: < 1.0 EU per 1 µg of protein (determined by LAL method) Preparation: Liquid purified protein Applications: Protocol: Activity Assay 1. Prepare a 1.4 ml assay buffer (Assay buffer: 58 mM glycylglycine, 2.0 mM adenosine 5'- diphosphate, 2.3 mM beta-nicotinamide adenine dinucleotide phosphate, 10 mM magnesium chloride, 10 mM glucose, 2 unit hexokinase, 1 unit glucose-6-phosphate dehydrogenase, 0.003% (w/v) BSA, pH 7.5.) - G-6-PDH/Hex (sigma,Cat.No. H-8629) 2. Add 50 ul of recombinant Adenylate kinase isoenzyme 2 protein with various concentrations (0.5 ug, 1 ug) and read the increase in A340nm for 5 minutes. Protein Description: Recombinant human AK2 protein, fused to His-tag at N-terminus, was expressed in E.coli and purified by using conventional chromatography techniques. -

Next-Generation Sequencing, FISH Mapping and Synteny- Based Modeling Reveal Mechanisms of Decreasing Dysploidy in Cucumis
The Plant Journal (2014) 77, 16–30 doi: 10.1111/tpj.12355 Next-generation sequencing, FISH mapping and synteny- based modeling reveal mechanisms of decreasing dysploidy in Cucumis Luming Yang1,†, Dal-Hoe Koo1,†, Dawei Li1,2, Tao Zhang1, Jiming Jiang1, Feishi Luan3, Susanne S. Renner4, Elizabeth Henaff 5, Walter Sanseverino6, Jordi Garcia-Mas6, Josep Casacuberta5, Douglas A. Senalik1,7, Philipp W. Simon1,7, Jinfeng Chen8 and Yiqun Weng1,7,* 1Horticulture Department, University of Wisconsin, Madison, WI 53706, USA, 2Horticulture College, Northwest A & F University, Yangling 712100, China, 3Horticulture College, Northeast Agricultural University, Harbin 150030, China, 4Department of Biology, University of Munich, 80638 Munich, Germany, 5Centre for Research in Agricultural Genomics Consejo Superior de Investigaciones Cientı´ficas-Institut de Recerca i Tecnolo- gia Agroalimenta` ries-Universitat Auto` noma de Barcelona-Universitat de Barcelona, 08193 Barcelona, Spain, 6Institut de Recerca i Tecnologia Agroalimenta` ries, Centre for Research in Agricultural Genomics Consejo Superior de Inves- tigaciones Cientı´ficas-Institut de Recerca i Tecnologia Agroalimenta` ries-Universitat Auto` noma de Barcelona-Universitat de Barcelona, 08193 Barcelona, Spain, 7US Department of Agriculture/Agricultural Research Service, Vegetable Crops Research Unit, 1575 Linden Drive, Madison, WI 53706, USA, and 8College of Horticulture, Nanjing Agricultural University, Nanjing 210095, China Received 27 July 2013; revised 7 October 2013; accepted 10 October 2013; published online 15 October 2013. *For correspondence (e-mail [email protected]). †These authors contributed equally to this work. SUMMARY In the large Cucurbitaceae genus Cucumis, cucumber (C. sativus) is the only species with 2n = 2x = 14 chro- mosomes. The majority of the remaining species, including melon (C.