Somatic Polyploidy Is Associated with the Upregulation of C-MYC Interacting Genes and EMT-Like Signature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

(HPG) Axis Published: 18 April 2017 Matthew D
www.nature.com/scientificreports OPEN Widespread patterns of sexually dimorphic gene expression in an avian hypothalamic–pituitary– Received: 30 November 2016 Accepted: 16 February 2017 gonadal (HPG) axis Published: 18 April 2017 Matthew D. MacManes1, Suzanne H. Austin2, Andrew S. Lang1, April Booth2, Victoria Farrar2 & Rebecca M. Calisi2 The hypothalamic-pituitary-gonadal (HPG) axis is a key biological system required for reproduction and associated sexual behaviors to occur. In the avian reproductive model of the rock dove (Columba livia), we characterized the transcript community of each tissue of the HPG axis in both sexes, thereby significantly expanding our mechanistic insight into HPG activity. We report greater sex-biased differential expression in the pituitary as compared to the hypothalamus, with multiple genes more highly expressed in the male pituitary being related to secretory function, and multiple genes more highly expressed in the female pituitary being related to reproduction, growth, and development. We report tissue-specific and sex-biased expression in genes commonly investigated when studying reproduction, highlighting the need for sex parity in future studies. In addition, we uncover new targets of investigation in both sexes, which could potentially change our understanding of HPG function. The hypothalamic-pituitary-gonadal (HPG) axis is a system comprised of endocrine glands whose function is vital to the regulation of reproduction and associated behaviors (Fig. 1). In all vertebrates studied, from humans to Agnatha, the jawless fishes, the HPG axis is present and its function is generally conserved1. For reproduc- tion to occur, the hypothalamus must produce and secrete gonadotropin-releasing hormone (GnRH), which causes the pituitary gland to secrete gonadotropins, luteinizing hormone (LH) and follicle stimulating hormone (FSH)2. -

Overexpression of Salicylic Acid Carboxyl Methyltransferase (Cssamt1) Enhances Tolerance to Huanglongbing Disease in Wanjincheng Orange (Citrus Sinensis (L.) Osbeck)
International Journal of Molecular Sciences Article Overexpression of Salicylic Acid Carboxyl Methyltransferase (CsSAMT1) Enhances Tolerance to Huanglongbing Disease in Wanjincheng Orange (Citrus sinensis (L.) Osbeck) Xiuping Zou * , Ke Zhao, Yunuo Liu, Meixia Du, Lin Zheng, Shuai Wang, Lanzhen Xu, Aihong Peng, Yongrui He, Qin Long and Shanchun Chen * Citrus Research Institute, Southwest University/Chinese Academy of Agricultural Sciences, Chongqing 400716, China; [email protected] (K.Z.); [email protected] (Y.L.); [email protected] (M.D.); [email protected] (L.Z.); [email protected] (S.W.); [email protected] (L.X.); [email protected] (A.P.); [email protected] (Y.H.); [email protected] (Q.L.) * Correspondence: [email protected] (X.Z.); [email protected] (S.C.) Abstract: Citrus Huanglongbing (HLB) disease or citrus greening is caused by Candidatus Liberibacter asiaticus (Las) and is the most devastating disease in the global citrus industry. Salicylic acid (SA) plays a central role in regulating plant defenses against pathogenic attack. SA methyltransferase (SAMT) modulates SA homeostasis by converting SA to methyl salicylate (MeSA). Here, we report on the functions of the citrus SAMT (CsSAMT1) gene from HLB-susceptible Wanjincheng orange Citation: Zou, X.; Zhao, K.; Liu, Y.; (Citrus sinensis (L.) Osbeck) in plant defenses against Las infection. The CsSAMT1 cDNA was Du, M.; Zheng, L.; Wang, S.; Xu, L.; expressed in yeast. Using in vitro enzyme assays, yeast expressing CsSAMT1 was confirmed to Peng, A.; He, Y.; Long, Q.; et al. specifically catalyze the formation of MeSA using SA as a substrate. Transgenic Wanjincheng orange Overexpression of Salicylic Acid plants overexpressing CsSAMT1 had significantly increased levels of SA and MeSA compared to Carboxyl Methyltransferase wild-type controls. -

The Role of E2F7 and E2F8 in Squamous Differentiation, UV- and Chemotherapy-Induced Responses in Keratinocytes
The role of E2F7 and E2F8 in squamous differentiation, UV- and chemotherapy-induced responses in keratinocytes Mehlika Hazar Rethinam MScBiotech (Hons) A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2014 UQ Diamantina Institute i Abstract Among keratinocyte-derived squamous cell carcinoma (SCC), cutaneous SCC (CSCC) is the second most common cutaneous cancer type and the most common of the potentially fatal skin cancers. Approximately 90% of head and neck cancers are SCC (HNSCC) and HNSCC worldwide is the sixth most common cancer type afflicting mankind. While resectable disease may be treated by surgery with radiation or chemoradiation, there are still no curative options for advanced, unresectable disease. However, chemotherapy alone may offer a hope for unresectable, disseminated SCC, where 50-60% of patients have disease recurrence within 2 years and approximately 30% of these develop metastatic disease. The mainstay of chemotherapeutic treatment in SCC is the platinum-based drug, cisplatin, 5-fluorouracil (5- FU), taxanes, and anti-EGFR monoclonal antibody such as Cetuximab. Nonetheless, despite advances in treatment techniques, the 5-year survival rate still remains at around 55%. Therefore, there remains a lack of options for recurrent or metastatic disease. The E2F family of transcription factors has emerged as key regulators of proliferation, differentiation and response to stress and apoptotic stimuli in keratinocytes. Consistent with these roles, dysregulation of E2F expression/activity is a common occurrence in cancer and SCC in particular. Thus, better understanding of the E2F family of proteins is essential to establish how these processes are disrupted during SCC genesis. -

E2f8 Mediates Tumor Suppression in Postnatal Liver Development
Downloaded from http://www.jci.org on January 18, 2017. https://doi.org/10.1172/JCI85506 The Journal of Clinical Investigation RESEARCH ARTICLE E2f8 mediates tumor suppression in postnatal liver development Lindsey N. Kent,1,2,3 Jessica B. Rakijas,1,2,3 Shusil K. Pandit,4 Bart Westendorp,4 Hui-Zi Chen,1,2,3,5 Justin T. Huntington,1,2,3,6 Xing Tang,1,2,3 Sooin Bae,1,2,3 Arunima Srivastava,1,2,3,7 Shantibhusan Senapati,1,2,3 Christopher Koivisto,1,2,3 Chelsea K. Martin,1,2,3 Maria C. Cuitino,1,2,3 Miguel Perez,1,2,3 Julian M. Clouse,1,2,3 Veda Chokshi,1,2,3 Neelam Shinde,1,2,3 Raleigh Kladney,1,2,3 Daokun Sun,1,2,3 Antonio Perez-Castro,1,2,3 Ramadhan B. Matondo,4 Sathidpak Nantasanti,4 Michal Mokry,8 Kun Huang,9 Raghu Machiraju,7 Soledad Fernandez,9 Thomas J. Rosol,10 Vincenzo Coppola,1,3 Kamal S. Pohar,11 James M. Pipas,12 Carl R. Schmidt,6 Alain de Bruin,4,13 and Gustavo Leone1,2,3 1Department of Cancer Biology and Genetics, College of Medicine, 2Department of Molecular Genetics, College of Biological Sciences, and 3Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio, USA. 4Department of Pathobiology, Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands. 5Division of Medical Oncology, Department of Internal Medicine, 6Department of Surgery, College of Medicine, and 7Department of Computer Science and Engineering, College of Engineering, The Ohio State University, Columbus, Ohio, USA. 8Department of Pediatrics, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Utrecht, Netherlands. -

Loss of Fam60a, a Sin3a Subunit, Results in Embryonic Lethality and Is Associated with Aberrant Methylation at a Subset of Gene
RESEARCH ARTICLE Loss of Fam60a, a Sin3a subunit, results in embryonic lethality and is associated with aberrant methylation at a subset of gene promoters Ryo Nabeshima1,2, Osamu Nishimura3,4, Takako Maeda1, Natsumi Shimizu2, Takahiro Ide2, Kenta Yashiro1†, Yasuo Sakai1, Chikara Meno1, Mitsutaka Kadota3,4, Hidetaka Shiratori1†, Shigehiro Kuraku3,4*, Hiroshi Hamada1,2* 1Developmental Genetics Group, Graduate School of Frontier Biosciences, Osaka University, Suita, Japan; 2Laboratory for Organismal Patterning, RIKEN Center for Developmental Biology, Kobe, Japan; 3Phyloinformatics Unit, RIKEN Center for Life Science Technologies, Kobe, Japan; 4Laboratory for Phyloinformatics, RIKEN Center for Biosystems Dynamics Research, Kobe, Japan Abstract We have examined the role of Fam60a, a gene highly expressed in embryonic stem cells, in mouse development. Fam60a interacts with components of the Sin3a-Hdac transcriptional corepressor complex, and most Fam60a–/– embryos manifest hypoplasia of visceral organs and die in utero. Fam60a is recruited to the promoter regions of a subset of genes, with the expression of these genes being either up- or down-regulated in Fam60a–/– embryos. The DNA methylation level of the Fam60a target gene Adhfe1 is maintained at embryonic day (E) 7.5 but markedly reduced at –/– *For correspondence: E9.5 in Fam60a embryos, suggesting that DNA demethylation is enhanced in the mutant. [email protected] (SK); Examination of genome-wide DNA methylation identified several differentially methylated regions, [email protected] (HH) which were preferentially hypomethylated, in Fam60a–/– embryos. Our data suggest that Fam60a is †These authors contributed required for proper embryogenesis, at least in part as a result of its regulation of DNA methylation equally to this work at specific gene promoters. -

(BPA) Exposure Biomarkers in Ovarian Cancer
Journal of Clinical Medicine Article Identification of Potential Bisphenol A (BPA) Exposure Biomarkers in Ovarian Cancer Aeman Zahra 1, Qiduo Dong 1, Marcia Hall 1,2 , Jeyarooban Jeyaneethi 1, Elisabete Silva 1, Emmanouil Karteris 1,* and Cristina Sisu 1,* 1 Biosciences, College of Health, Medicine and Life Sciences, Brunel University London, Uxbridge UB8 3PH, UK; [email protected] (A.Z.); [email protected] (Q.D.); [email protected] (M.H.); [email protected] (J.J.); [email protected] (E.S.) 2 Mount Vernon Cancer Centre, Northwood HA6 2RN, UK * Correspondence: [email protected] (E.K.); [email protected] (C.S.) Abstract: Endocrine-disrupting chemicals (EDCs) can exert multiple deleterious effects and have been implicated in carcinogenesis. The xenoestrogen Bisphenol A (BPA) that is found in various consumer products has been involved in the dysregulation of numerous signalling pathways. In this paper, we present the analysis of a set of 94 genes that have been shown to be dysregulated in presence of BPA in ovarian cancer cell lines since we hypothesised that these genes might be of biomarker potential. This study sought to identify biomarkers of disease and biomarkers of disease- associated exposure. In silico analyses took place using gene expression data extracted from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) databases. Differential expression was further validated at protein level using immunohistochemistry on an ovarian cancer tissue microarray. We found that 14 out of 94 genes are solely dysregulated in the presence of BPA, while the remaining 80 genes are already dysregulated (p-value < 0.05) in their expression pattern Citation: Zahra, A.; Dong, Q.; Hall, as a consequence of the disease. -
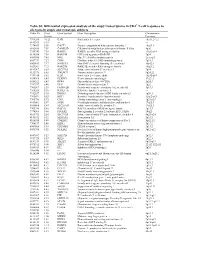
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

Downloaded the “Top Edge” Version
bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Drosophila models of pathogenic copy-number variant genes show global and 2 non-neuronal defects during development 3 Short title: Non-neuronal defects of fly homologs of CNV genes 4 Tanzeen Yusuff1,4, Matthew Jensen1,4, Sneha Yennawar1,4, Lucilla Pizzo1, Siddharth 5 Karthikeyan1, Dagny J. Gould1, Avik Sarker1, Yurika Matsui1,2, Janani Iyer1, Zhi-Chun Lai1,2, 6 and Santhosh Girirajan1,3* 7 8 1. Department of Biochemistry and Molecular Biology, Pennsylvania State University, 9 University Park, PA 16802 10 2. Department of Biology, Pennsylvania State University, University Park, PA 16802 11 3. Department of Anthropology, Pennsylvania State University, University Park, PA 16802 12 4 contributed equally to work 13 14 *Correspondence: 15 Santhosh Girirajan, MBBS, PhD 16 205A Life Sciences Building 17 Pennsylvania State University 18 University Park, PA 16802 19 E-mail: [email protected] 20 Phone: 814-865-0674 21 1 bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 22 ABSTRACT 23 While rare pathogenic copy-number variants (CNVs) are associated with both neuronal and non- 24 neuronal phenotypes, functional studies evaluating these regions have focused on the molecular 25 basis of neuronal defects. -

Duplication 9P and Their Implication to Phenotype
Guilherme et al. BMC Medical Genetics (2014) 15:142 DOI 10.1186/s12881-014-0142-1 RESEARCH ARTICLE Open Access Duplication 9p and their implication to phenotype Roberta Santos Guilherme1, Vera Ayres Meloni1, Ana Beatriz Alvarez Perez1, Ana Luiza Pilla1, Marco Antonio Paula de Ramos1, Anelisa Gollo Dantas1, Sylvia Satomi Takeno1, Leslie Domenici Kulikowski2 and Maria Isabel Melaragno1* Abstract Background: Trisomy 9p is one of the most common partial trisomies found in newborns. We report the clinical features and cytogenomic findings in five patients with different chromosome rearrangements resulting in complete 9p duplication, three of them involving 9p centromere alterations. Methods: The rearrangements in the patients were characterized by G-banding, SNP-array and fluorescent in situ hybridization (FISH) with different probes. Results: Two patients presented de novo dicentric chromosomes: der(9;15)t(9;15)(p11.2;p13) and der(9;21)t(9;21) (p13.1;p13.1). One patient presented two concomitant rearranged chromosomes: a der(12)t(9;12)(q21.13;p13.33) and an psu i(9)(p10) which showed FISH centromeric signal smaller than in the normal chromosome 9. Besides the duplication 9p24.3p13.1, array revealed a 7.3 Mb deletion in 9q13q21.13 in this patient. The break in the psu i(9) (p10) probably occurred in the centromere resulting in a smaller centromere and with part of the 9q translocated to the distal 12p with the deletion 9q occurring during this rearrangement. Two patients, brother and sister, present 9p duplication concomitant to 18p deletion due to an inherited der(18)t(9;18)(p11.2;p11.31)mat. -

The Atypical E2F Family Member E2F7 Couples the P53 and RB Pathways During Cellular Senescence
Downloaded from genesdev.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence Ozlem Aksoy,1,2,3,6 Agustin Chicas,1,3,6 Tianying Zeng,4 Zhen Zhao,1,3 Mila McCurrach,3 Xiaowo Wang,4 and Scott W. Lowe1,3,5,7 1Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA; 2Watson School of Biological Sciences, 3Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA; 4MOE Key Laboratory of Bioinformatics, Bioinformatics Division, TNLIST, Department of Automation, Tsinghua University, Beijing 100084, China; 5Howard Hughes Medical Institute, New York, New York 10065, USA Oncogene-induced senescence is an anti-proliferative stress response program that acts as a fail-safe mechanism to limit oncogenic transformation and is regulated by the retinoblastoma protein (RB) and p53 tumor suppressor pathways. We identify the atypical E2F family member E2F7 as the only E2F transcription factor potently up- regulated during oncogene-induced senescence, a setting where it acts in response to p53 as a direct transcriptional target. Once induced, E2F7 binds and represses a series of E2F target genes and cooperates with RB to efficiently promote cell cycle arrest and limit oncogenic transformation. Disruption of RB triggers a further increase in E2F7, which induces a second cell cycle checkpoint that prevents unconstrained cell division despite aberrant DNA replication. Mechanistically, E2F7 compensates for the loss of RB in repressing mitotic E2F target genes. Together, our results identify a causal role for E2F7 in cellular senescence and uncover a novel link between the RB and p53 pathways. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing