Design and Synthesis of Novel Antimalarial Agents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information
Supplementary Information Table S1. Categories of transcripts significantly regulated in salt-stressed Malus zumi. Number Expression a Putative Annotation Genome Genebank Identities p-value b locus Accession Signal transduction Kinase leucine-rich repeat transmembrane protein 5.41 × 10-5 1 I Chr 15 NP_199948 64% kinase 1 S leucine-rich repeat family protein kinase Chr 15 NP_179336 42% 1.02 × 10-4 1 I tousled-like serine/threonine kinase Chr 11 NP_568405 82% 3.20 × 10-5 1 I CIPK5 Chr 1 NP_568241 79% 7.74 × 10-5 1 S CIPK6 Chr 2 NP_194825 80% 1.98 × 10-7 1 S protein kinase family protein Chr 6 NP_194952 50% 2.25 × 10-5 Transcription factor 1 I IAA-LEUCINE RESISTANT3 Chr 3 NP_200279 89% 4.97 × 10-4 1 I IAA26 Chr 15 NP_188271 80% 3.58 × 10-6 1 S GT-like trihelix DNA-binding protein Chr 15 NP_177814 37% 3.04 × 10-6 1 S zinc finger (CCCH-type) family protein Chr 11 NP_200670 59% 8.13 × 10-5 1 I WRKY family transcription factor Chr 12 NP_001078015 50% 4.52 × 10-5 1 S GRAS family transcription factor Chr 10 XP_002322514 52% 3.58 × 10-4 2 S AP2 transcription factor Chr 15 NP_173355 70% 4.26 × 10-6 1 I SALT TOLERANCE homolog protein Chr 5 NP_849598 68% 5.37 × 10-7 1 S Auxin response factor Chr 7 NP_182176 70% 2.78 × 10-3 Int. J. Mol. Sci. 2013, 14 S2 Table S1. Cont. Number Expression a Putative Annotation Genome Genebank Identities p-value b locus Accession ROS elimination 1 S glutathione transferase Chr 3 NP_850479 62% 3.17 × 10-8 1 S peroxidase Chr 2 NP_201440 71% 4.26 × 10-3 1 S peroxidase Chr 10 NP_197022 54% 3.81 × 10-7 1 S peroxidase Chr 10 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Spermidine Synthase (SPDS) Undergoes Concerted Structural Rearrangements Upon Ligand Binding – a Case Study of the Two SPDS Isoforms from Arabidopsis Thaliana
fpls-10-00555 May 4, 2019 Time: 16:20 # 1 ORIGINAL RESEARCH published: 07 May 2019 doi: 10.3389/fpls.2019.00555 Spermidine Synthase (SPDS) Undergoes Concerted Structural Rearrangements Upon Ligand Binding – A Case Study of the Two SPDS Isoforms From Arabidopsis thaliana Bartosz Sekula* and Zbigniew Dauter Synchrotron Radiation Research Section, Macromolecular Crystallography Laboratory, National Cancer Institute, Argonne, IL, United States Spermidine synthases (SPDSs) catalyze the production of the linear triamine, spermidine, from putrescine. They utilize decarboxylated S-adenosylmethionine (dc- SAM), a universal cofactor of aminopropyltransferases, as a donor of the aminopropyl Edited by: Antonio F. Tiburcio, moiety. In this work, we describe crystal structures of two SPDS isoforms from University of Barcelona, Spain Arabidopsis thaliana (AtSPDS1 and AtSPDS2). AtSPDS1 and AtSPDS2 are dimeric Reviewed by: enzymes that share the fold of the polyamine biosynthesis proteins. Subunits of both Taku Takahashi, Okayama University, Japan isoforms present the characteristic two-domain structure. Smaller, N-terminal domain Miguel A. Blazquez, is built of the two b-sheets, while the C-terminal domain has a Rossmann fold-like Spanish National Research Council topology. The catalytic cleft composed of two main compartments, the dc-SAM binding (CSIC), Spain site and the polyamine groove, is created independently in each AtSPDS subunits at *Correspondence: Bartosz Sekula the domain interface. We also provide the structural details about the dc-SAM binding [email protected]; mode and the inhibition of SPDS by a potent competitive inhibitor, cyclohexylamine [email protected] (CHA). CHA occupies the polyamine binding site of AtSPDS where it is bound at the Specialty section: bottom of the active site with the amine group placed analogously to the substrate. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -
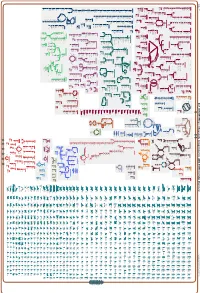
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -

Supplementary Table 3 Gene Microarray Analysis: PRL+E2 Vs
Supplementary Table 3 Gene microarray analysis: PRL+E2 vs. control ID1 Field1 ID Symbol Name M Fold P Value 69 15562 206115_at EGR3 early growth response 3 2,36 5,13 4,51E-06 56 41486 232231_at RUNX2 runt-related transcription factor 2 2,01 4,02 6,78E-07 41 36660 227404_s_at EGR1 early growth response 1 1,99 3,97 2,20E-04 396 54249 36711_at MAFF v-maf musculoaponeurotic fibrosarcoma oncogene homolog F 1,92 3,79 7,54E-04 (avian) 42 13670 204222_s_at GLIPR1 GLI pathogenesis-related 1 (glioma) 1,91 3,76 2,20E-04 65 11080 201631_s_at IER3 immediate early response 3 1,81 3,50 3,50E-06 101 36952 227697_at SOCS3 suppressor of cytokine signaling 3 1,76 3,38 4,71E-05 16 15514 206067_s_at WT1 Wilms tumor 1 1,74 3,34 1,87E-04 171 47873 238623_at NA NA 1,72 3,30 1,10E-04 600 14687 205239_at AREG amphiregulin (schwannoma-derived growth factor) 1,71 3,26 1,51E-03 256 36997 227742_at CLIC6 chloride intracellular channel 6 1,69 3,23 3,52E-04 14 15038 205590_at RASGRP1 RAS guanyl releasing protein 1 (calcium and DAG-regulated) 1,68 3,20 1,87E-04 55 33237 223961_s_at CISH cytokine inducible SH2-containing protein 1,67 3,19 6,49E-07 78 32152 222872_x_at OBFC2A oligonucleotide/oligosaccharide-binding fold containing 2A 1,66 3,15 1,23E-05 1969 32201 222921_s_at HEY2 hairy/enhancer-of-split related with YRPW motif 2 1,64 3,12 1,78E-02 122 13463 204015_s_at DUSP4 dual specificity phosphatase 4 1,61 3,06 5,97E-05 173 36466 227210_at NA NA 1,60 3,04 1,10E-04 117 40525 231270_at CA13 carbonic anhydrase XIII 1,59 3,02 5,62E-05 81 42339 233085_s_at OBFC2A oligonucleotide/oligosaccharide-binding -

Norspermine Substitutes for Thermospermine in the Control of Stem Elongation in Arabidopsis Thaliana
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 584 (2010) 3042–3046 journal homepage: www.FEBSLetters.org Norspermine substitutes for thermospermine in the control of stem elongation in Arabidopsis thaliana Jun-Ichi Kakehi a, Yoshitaka Kuwashiro a, Hiroyasu Motose a, Kazuei Igarashi b, Taku Takahashi a,* a Graduate School of Natural Science and Technology, Okayama University, Okayama 700-8530, Japan b Graduate School of Pharmaceutical Sciences, Chiba University, Chiba 260-8675, Japan article info abstract Article history: Thermospermine is a structural isomer of spermine and is required for stem elongation in Arabid- Received 26 March 2010 opsis thaliana. We noted the C3C3 arrangement of carbon chains in thermospermine (C3C3C4), Revised 16 May 2010 which is not present in spermine (C3C4C3), and examined if it is functionally replaced with norsper- Accepted 17 May 2010 mine (C3C3C3) or not. Exogenous application of norspermine to acl5, a mutant defective in the syn- Available online 24 May 2010 thesis of thermospermine, partially suppressed its dwarf phenotype, and down-regulated the level Edited by Ulf-Ingo Flügge of the acl5 transcript which is much higher than that of the ACL5 transcript in the wild type. Further- more, in the Zinnia culture, differentiation of mesophyll cells into tracheary elements was blocked by thermospermine and norspermine but not by spermine. Our results indicate that norspermine Keywords: Arabidopsis can functionally substitute for thermospermine. Thermospermine Ó 2010 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved. Norspermine Polyamine Stem elongation Xylem 1. -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Polyamines and Their Biosynthesis/Catabolism Genes Are Differentially Modulated in Response to Heat Versus Cold Stress in Tomato Leaves (Solanum Lycopersicum L.)
cells Article Polyamines and Their Biosynthesis/Catabolism Genes Are Differentially Modulated in Response to Heat Versus Cold Stress in Tomato Leaves (Solanum lycopersicum L.) Rakesh K. Upadhyay 1,2 , Tahira Fatima 2, Avtar K. Handa 2 and Autar K. Mattoo 1,* 1 Sustainable Agricultural Systems Laboratory, United States Department of Agriculture, Agricultural Research Service, Henry A. Wallace Beltsville Agricultural Research Center, Beltsville, MD 20705-2350, USA; [email protected] 2 Center of Plant Biology, Department of Horticulture and Landscape Architecture, Purdue University, W. Lafayette, IN 47907, USA; [email protected] (T.F.); [email protected] (A.K.H.) * Correspondence: [email protected]; Tel.: +1-301-504-6622 Received: 20 June 2020; Accepted: 20 July 2020; Published: 22 July 2020 Abstract: Polyamines (PAs) regulate growth in plants and modulate the whole plant life cycle. They have been associated with different abiotic and biotic stresses, but little is known about the molecular regulation involved. We quantified gene expression of PA anabolic and catabolic pathway enzymes in tomato (Solanum lycopersicum cv. Ailsa Craig) leaves under heat versus cold stress. These include arginase 1 and 2, arginine decarboxylase 1 and 2, agmatine iminohydrolase/deiminase 1, N-carbamoyl putrescine amidase, two ornithine decarboxylases, three S-adenosylmethionine decarboxylases, two spermidine synthases; spermine synthase; flavin-dependent polyamine oxidases (SlPAO4-like and SlPAO2) and copper dependent amine oxidases (SlCuAO and SlCuAO-like). The spatiotemporal transcript abundances using qRT-PCR revealed presence of their transcripts in all tissues examined, with higher transcript levels observed for SAMDC1, SAMDC2 and ADC2 in most tissues. Cellular levels of free and conjugated forms of putrescine and spermidine were found to decline during heat stress while they increased in response to cold stress, revealing their differential responses. -

Homospermidine, Spermidine, and Putrescine: The
AN ABSTRACT OF THE THESIS OF Paula Allene Tower for the degree of Doctor of Philosophy in Microbiology presented on July 28, 1987. Title: Homospermidine, Spermidine, and Putrescine: The Biosynthesis and Metabolism of Polyamines in Rhizobium meliloti Redacted for privacy Abstract approved: Dr.4dolph J. Ferro Rhizobium, in symbiotic association with leguminous plants, is able to fix atmospheric nitrogen after first forming root nodules. Since polyamines are associated with and found in high concentration in rapidly growing cells and are thought to be important foroptimal cell growth, it is possible that these polycations are involved in the extensive cell proliferation characteristic of the nodulation process. As a first step towards elucidating the role(s) of poly- amines in the rhizobial-legume interaction, I have characterized polyamine biosynthesis and metabolism in free-living Rhizobium meliloti. In addition to the detection of putrescine and spermidine, the presence of a less common polyamine, homospermidine, wasconfirmed in Rhizobium meliloti, with homospermidine comprising 79 percent of the free polyamines in this procaryote. The presence of an exogenous polyamine both affected the intra- cellular levels of each polyamine pool and inhibited the accumula- tion of a second amine. DL-a-Difluoromethylornithine (DFMO), an irreversible inhibitor of ornithine decarboxylase, wasfound to: (1) inhibit the rhizobial enzyme both in vitro and in vivo; (2) increase the final optical density; and (3) create ashift in the dominant polyamine from homospermidine to spermidine. A series of radiolabeled amino acids and polyamines were studied as polyamine precursors. Ornithine, arginine, aspartic acid, putrescine, and spermidine, but not methionine, resulted in the isolation of labeled putrescine, spermidine, andhomospermidine. -

Polyamines Mitigate Antibiotic Inhibition of A.Actinomycetemcomitans Growth
Polyamines Mitigate Antibiotic Inhibition of A.actinomycetemcomitans Growth THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Allan Wattimena Graduate Program in Dentistry The Ohio State University 2017 Master's Examination Committee: Dr John Walters, Advisor Dr Purnima Kumar Dr Sara Palmer Dr Shareef Dabdoub Copyright by Allan Wattimena 2017 Abstract Polyamines are ubiquitous polycationic molecules that are present in all prokaryotic and eukaryotic cells. They are the breakdown products of amino acids and are important modulators of cell growth, stress and cell proliferation. Polyamines are present in higher concentrations in the periodontal pocket and may affect antibiotic resistance of bacterial biofilms. The effect of polyamines was investigated with amoxicillin (AMX), azithromycin (AZM) and doxycycline (DOX) on the growth of Aggregatibacter actinomycetemcomitans (A.a.) Y4 strain. Bacteria were grown in brain heart infusion broth under the following conditions: 1) A.a. only, 2) A.a. + antibiotic, 3) A.a. + antibiotic + polyamine mix (1.4mM putrescine, 0.4mM spermidine, 0.4mM spermine). Growth curve analysis, MIC determination and metatranscriptomic analysis were carried out. The presence of exogenous polyamines produced a small, but significant increase in growth of A.a. Polyamines mitigated the inhibitory effect of AMX, AZM and DOX on A.a. growth. Metatranscriptomic analysis revealed differing transcriptomic profiles when comparing AMX and AZM in the presence of polyamines. Polyamines produced a transient mitigation of AMX inhibition, but did not have a significant effect on gene transcription. Many gene transcription changes were seen when polyamines were in the presence of AZM.