Connecting Genes to Molecules: Identifying Small Molecules from Phytopathogenic Bacteria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Acidic Phospholipids,47, 58 Acidification Steps, 23 Adsorbent, 53
Index acidic phospholipids,47, 58 ATPase, 22, 107 acidification steps, 23 auto-oxidation, 48,49 adsorbent, 53 aequorin, 95 aggregated mitochondria, 23 bacterial cells, 10 alcohols, 49 bacteriorhodopsin (BR), 218,232 alkaline hydrolysis, I bee venom, 121 alkyl esters, 57, 59 binders,53 amide bond, 129 silica gel, 53, 65, 176 amide linkage, 14 silica gel H, 176 amino alcohol, II bioactive phospholipids, 144 amino-group labelling reagents, 121 blanching, 50 aminopeptidase, 17 buoyant density, 16 aminophospholipid, 113, 118 butyl hydroxy toluene (BHT), 49,212 aminophospholipid pump, 119, 140 aminophospholipid translocase, 11 3 amphotericin B (AmB), 107 Cal+ -ATPases, 28, 107 N-(1-deoxyD-fructos-1-yl) AmB, 107 Cal+ -uptake, 27 anchored lipids, 38 campesterol, 104 angular amplitude, 89 calciferol, 78 anilino-8-naphthalene sulfonate (ANS), Candida albicans, 59, 60, 61, 76, 78, 102, 94, 103 104, 108 animal cells, 10 caproic acid, 129, 130 animal tissues, 9 carotenoids, 37 anion transporter, 135 cell debris, 33 anisotropy, 81 , 89, 95, 96, 97, 102, 104, cell disintegrator, 19 105 ceramide, 14, 129, 194 antagonists, 178 ceramide monohexosides ( CMH), 60 antioxidant, 49, 50 cerebroside, 14 apolar lipids, 56, 57, 69 chemical probes,112 arachidonic acid (AA), 144,153, 161 chloroplast, 32, 33 artificial membrane, 83 isolation, 32, 33 ascending chromatography, 54 purification, 33 asymmetric topology, 128 cholesterol, 14, 15,212 asymmetry, 112, 113,119 choline, 7, 9 atebrin, 95 chromatograms, 55 Index 249 chromatographic analyses, 52 ELISA, 146, 148 -

Bioluminescent Properties of Semi-Synthetic Obelin and Aequorin Activated by Coelenterazine Analogues with Modifications of C-2, C-6, and C-8 Substituents
International Journal of Molecular Sciences Article Bioluminescent Properties of Semi-Synthetic Obelin and Aequorin Activated by Coelenterazine Analogues with Modifications of C-2, C-6, and C-8 Substituents 1, 2,3, 1 2, Elena V. Eremeeva y , Tianyu Jiang y , Natalia P. Malikova , Minyong Li * and Eugene S. Vysotski 1,* 1 Photobiology Laboratory, Institute of Biophysics SB RAS, Federal Research Center “Krasnoyarsk Science Center SB RAS”, Krasnoyarsk 660036, Russia; [email protected] (E.V.E.); [email protected] (N.P.M.) 2 Key Laboratory of Chemical Biology (MOE), Department of Medicinal Chemistry, School of Pharmaceutical Sciences, Shandong University, Jinan 250012, China; [email protected] 3 State Key Laboratory of Microbial Technology, Shandong University–Helmholtz Institute of Biotechnology, Shandong University, Qingdao 266237, China * Correspondence: [email protected] (M.L.); [email protected] (E.S.V.); Tel.: +86-531-8838-2076 (M.L.); +7-(391)-249-44-30 (E.S.V.); Fax: +86-531-8838-2076 (M.L.); +7-(391)-290-54-90 (E.S.V.) These authors contributed equally to this work. y Received: 23 June 2020; Accepted: 27 July 2020; Published: 30 July 2020 Abstract: Ca2+-regulated photoproteins responsible for bioluminescence of a variety of marine organisms are single-chain globular proteins within the inner cavity of which the oxygenated coelenterazine, 2-hydroperoxycoelenterazine, is tightly bound. Alongside with native coelenterazine, photoproteins can also use its synthetic analogues as substrates to produce flash-type bioluminescence. However, information on the effect of modifications of various groups of coelenterazine and amino acid environment of the protein active site on the bioluminescent properties of the corresponding semi-synthetic photoproteins is fragmentary and often controversial. -

Supplementary Information
Supplementary Information Table S1. Categories of transcripts significantly regulated in salt-stressed Malus zumi. Number Expression a Putative Annotation Genome Genebank Identities p-value b locus Accession Signal transduction Kinase leucine-rich repeat transmembrane protein 5.41 × 10-5 1 I Chr 15 NP_199948 64% kinase 1 S leucine-rich repeat family protein kinase Chr 15 NP_179336 42% 1.02 × 10-4 1 I tousled-like serine/threonine kinase Chr 11 NP_568405 82% 3.20 × 10-5 1 I CIPK5 Chr 1 NP_568241 79% 7.74 × 10-5 1 S CIPK6 Chr 2 NP_194825 80% 1.98 × 10-7 1 S protein kinase family protein Chr 6 NP_194952 50% 2.25 × 10-5 Transcription factor 1 I IAA-LEUCINE RESISTANT3 Chr 3 NP_200279 89% 4.97 × 10-4 1 I IAA26 Chr 15 NP_188271 80% 3.58 × 10-6 1 S GT-like trihelix DNA-binding protein Chr 15 NP_177814 37% 3.04 × 10-6 1 S zinc finger (CCCH-type) family protein Chr 11 NP_200670 59% 8.13 × 10-5 1 I WRKY family transcription factor Chr 12 NP_001078015 50% 4.52 × 10-5 1 S GRAS family transcription factor Chr 10 XP_002322514 52% 3.58 × 10-4 2 S AP2 transcription factor Chr 15 NP_173355 70% 4.26 × 10-6 1 I SALT TOLERANCE homolog protein Chr 5 NP_849598 68% 5.37 × 10-7 1 S Auxin response factor Chr 7 NP_182176 70% 2.78 × 10-3 Int. J. Mol. Sci. 2013, 14 S2 Table S1. Cont. Number Expression a Putative Annotation Genome Genebank Identities p-value b locus Accession ROS elimination 1 S glutathione transferase Chr 3 NP_850479 62% 3.17 × 10-8 1 S peroxidase Chr 2 NP_201440 71% 4.26 × 10-3 1 S peroxidase Chr 10 NP_197022 54% 3.81 × 10-7 1 S peroxidase Chr 10 -

Bioluminescence Related Publications from JNC Corporation
Bioluminescence related publications since 1985 110) Inouye, S. and Hojo, H. (2018) Revalidation of recombinant aequorin as a light emission standard: Estimation of specific activity of Gaussia luciferase. Biochem. Biophys. Res. Commun. 507: 242-245. 109) Yokawa, S., Suzuki, T., Hayashi, A., Inouye, S., Inoh, Y. and Furuno, T. (2018) Video-rate bioluminescence imaging of degranulation of mast cells attached to the extracellular matrix. Front. Cell Dev. Biol. 6: 74. 108) Inouye, S., Tomabechi, Y., Hosoya, T., Sekine, S. and Shirouzu, M. (2018) Slow luminescence kinetics of semi-synthetic aequorin:expression, purification and structure determination of cf3-aequorin. J. Biochem. 164(3): 247–255. 107) Inouye, S. (2018) Single-step purification of recombinant Gaussia luciferase from serum-containing culture medium of mammalian cells. Protein Expr. Purif. 141: 32-38. 106) Inouye, S. and Sahara-Miura, Y. (2017) A fusion protein of the synthetic IgG-binding domain and aequorin: Expression and purification from E. coli cells and its application. Protein Expr. Purif. 137: 58-63. 105) Suzuki, T., Kanamori, T. and Inouye, S. (2017) Quantitative visualization of synchronized insulin secretion from 3D-cultured cells. Biochem. Biophys. Res. Commun. 486: 886-892. 104) Yokawa, S., Suzuki, T., Inouye, S., Inoh, Y., Suzuki, R., Kanamori, T., Furuno, T. and Hirashima, N. (2017) Visualization of glucagon secretion from pancreatic α cells by bioluminescence video microscopy: Identification of secretion sites in the intercellular contact regions. Biochem. Biophys. Res. Commun. 485: 725-730. 103) Inouye, S. and Suzuki, T. (2016) Protein expression of preferred human codon-optimized Gaussia luciferase genes with an artificial open-reading frame in mammalian and bacterial cells. -

BPS Complete Catalog
BPSCATALOG ENZYMES KITS CELL LINES SCREENING SERVICES INNOVATIVE PRODUCTS TO FUEL YOUR EXPERIMENTS Unique, expert portfolio 2017 OUR MISSION: To provide the highest quality life science products and services worldwide in a timely manner to assist in accelerating drug discovery research and development for the treatment of human diseases. INNOVATION WE CONTINUOUSLY STRIVE TO BE FIRST TO MARKET BY PROVIDING EXPERTISE IN EXPRESSING HIGHLY ACTIVE ENZYMES MULTIPLE ASSAY DETECTION FORMATS HUMAN, MURINE AND MONKEY VERSIONS OF RECOMBINANT PROTEINS BIOTINYLATED VERSIONS OF MANY IMMUNE CHECKPOINT RECEPTORS 1ST AND MOST EXTENSIVE PARP ISOZYME PORTFOLIO 1ST AND MOST EXTENSIVE PDE ISOZYME PORTFOLIO 1ST COMPLETE SUITE OF HDAC AND SIRT ENZYMES 1ST AVAILABLE PARP AND TANKYRASE PROFILING SERVICES 1ST COMMERCIALLY AVAILABLE HISTONE DEMETHYLASES LARGEST OFFERING OF METHYLTRANSFERASES LARGEST OFFERING OF DEMETHYLASES OVER 200 PRODUCTS AND SERVICES EXCLUSIVE TO BPS RELIABILITY BPS IS CITED IN PEER-REVIEWED JOURNALS AND PUBLICATIONS AROUND THE WORLD NATURE GENETICS THE JOURNAL OF BIOLOGICAL CHEMISTRY JOURNAL OF CELL SCIENCE ANALYTICAL BIOCHEMISTRY BBRC NATURE CHEMICAL BIOLOGY ACS MEDICINAL CHEMISTRY LETTERS JOURNAL OF NATURAL PRODUCTS CHEMMEDCHEM MOLECULAR CANCER THERAPEUTICS & MANY MORE TABLE OF CONTENTS ACETYLTRANSFERASE 4 APOPTOSIS 4-5 ANTIBODIES 6-9 BIOTINYLATION 10-11 BROMODOMAINS 12-14 CELL BASED ASSAY KITS 15 CELL LINES 16-17 CELL SURFACE RECEPTORS 18-20 CYTOKINE(S) 21-24 DEACETYLASE(S) 25-27 DEMETHYLASE(S) 28-29 HEAT SHOCK PROTEINS 30 IMMUNOTHERAPY -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Spermidine Synthase (SPDS) Undergoes Concerted Structural Rearrangements Upon Ligand Binding – a Case Study of the Two SPDS Isoforms from Arabidopsis Thaliana
fpls-10-00555 May 4, 2019 Time: 16:20 # 1 ORIGINAL RESEARCH published: 07 May 2019 doi: 10.3389/fpls.2019.00555 Spermidine Synthase (SPDS) Undergoes Concerted Structural Rearrangements Upon Ligand Binding – A Case Study of the Two SPDS Isoforms From Arabidopsis thaliana Bartosz Sekula* and Zbigniew Dauter Synchrotron Radiation Research Section, Macromolecular Crystallography Laboratory, National Cancer Institute, Argonne, IL, United States Spermidine synthases (SPDSs) catalyze the production of the linear triamine, spermidine, from putrescine. They utilize decarboxylated S-adenosylmethionine (dc- SAM), a universal cofactor of aminopropyltransferases, as a donor of the aminopropyl Edited by: Antonio F. Tiburcio, moiety. In this work, we describe crystal structures of two SPDS isoforms from University of Barcelona, Spain Arabidopsis thaliana (AtSPDS1 and AtSPDS2). AtSPDS1 and AtSPDS2 are dimeric Reviewed by: enzymes that share the fold of the polyamine biosynthesis proteins. Subunits of both Taku Takahashi, Okayama University, Japan isoforms present the characteristic two-domain structure. Smaller, N-terminal domain Miguel A. Blazquez, is built of the two b-sheets, while the C-terminal domain has a Rossmann fold-like Spanish National Research Council topology. The catalytic cleft composed of two main compartments, the dc-SAM binding (CSIC), Spain site and the polyamine groove, is created independently in each AtSPDS subunits at *Correspondence: Bartosz Sekula the domain interface. We also provide the structural details about the dc-SAM binding [email protected]; mode and the inhibition of SPDS by a potent competitive inhibitor, cyclohexylamine [email protected] (CHA). CHA occupies the polyamine binding site of AtSPDS where it is bound at the Specialty section: bottom of the active site with the amine group placed analogously to the substrate. -

Platelet-Neutrophil Interaction
PLATELET-NEUTROPHIL INTERACTION: NEUTROPHILS MODULATE PLATELET FUNCTION Richard William FAINT 1992 A thesis submitted to the University of London for the degree of Ph.D The Department of Haematology University College and Middlesex School of Medicine University College London 98 Chenies Mews London WC1E 6HX ProQuest Number: U049250 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest U049250 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 2 ABSTRACT Platelets are vital for the arrest of bleeding following tissue injury. Neutrophils play an important role in host defence against bacterial infection. In the milieu of the acute inflammatory response both cellular and non-cellular elements may interact to modify behaviour. Evidence suggests that leukocytes may play an active role in the modulation of platelet function. This interaction may be abnormal in certain pathological states. This study examined the effect of purified neutrophils upon both washed platelets and platelet-rich plasma, as well as in whole blood in vitro. Neutrophils were found to alter platelet behaviour by several mechanisms. These included transcellular metabolism of eicosanoids. -

Antibody List
產品編號 產品名稱 PA569955 1110059E24Rik Polyclonal Antibody PA569956 1110059E24Rik Polyclonal Antibody PA570131 1190002N15Rik Polyclonal Antibody 01-1234-42 123count eBeads Counting Beads MA512242 14.3.3 Pan Monoclonal Antibody (CG15) LFMA0074 14-3-3 beta Monoclonal Antibody (60C10) LFPA0077 14-3-3 beta Polyclonal Antibody PA137002 14-3-3 beta Polyclonal Antibody PA14647 14-3-3 beta Polyclonal Antibody PA515477 14-3-3 beta Polyclonal Antibody PA517425 14-3-3 beta Polyclonal Antibody PA522264 14-3-3 beta Polyclonal Antibody PA529689 14-3-3 beta Polyclonal Antibody MA134561 14-3-3 beta/epsilon/zeta Monoclonal Antibody (3C8) MA125492 14-3-3 beta/zeta Monoclonal Antibody (22-IID8B) MA125665 14-3-3 beta/zeta Monoclonal Antibody (4E2) 702477 14-3-3 delta/zeta Antibody (1H9L19), ABfinity Rabbit Monoclonal 711507 14-3-3 delta/zeta Antibody (1HCLC), ABfinity Rabbit Oligoclonal 702241 14-3-3 epsilon Antibody (5H10L5), ABfinity Rabbit Monoclonal 711273 14-3-3 epsilon Antibody (5HCLC), ABfinity Rabbit Oligoclonal PA517104 14-3-3 epsilon Polyclonal Antibody PA528937 14-3-3 epsilon Polyclonal Antibody PA529773 14-3-3 epsilon Polyclonal Antibody PA575298 14-3-3 eta (Lys81) Polyclonal Antibody MA524792 14-3-3 eta Monoclonal Antibody PA528113 14-3-3 eta Polyclonal Antibody PA529774 14-3-3 eta Polyclonal Antibody PA546811 14-3-3 eta Polyclonal Antibody MA116588 14-3-3 gamma Monoclonal Antibody (HS23) MA116587 14-3-3 gamma Monoclonal Antibody (KC21) PA529690 14-3-3 gamma Polyclonal Antibody PA578233 14-3-3 gamma Polyclonal Antibody 510700 14-3-3 Pan Polyclonal -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -
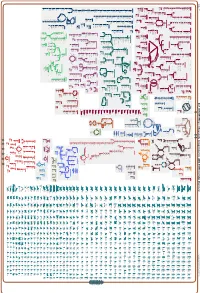
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -

Supplementary Table 3 Gene Microarray Analysis: PRL+E2 Vs
Supplementary Table 3 Gene microarray analysis: PRL+E2 vs. control ID1 Field1 ID Symbol Name M Fold P Value 69 15562 206115_at EGR3 early growth response 3 2,36 5,13 4,51E-06 56 41486 232231_at RUNX2 runt-related transcription factor 2 2,01 4,02 6,78E-07 41 36660 227404_s_at EGR1 early growth response 1 1,99 3,97 2,20E-04 396 54249 36711_at MAFF v-maf musculoaponeurotic fibrosarcoma oncogene homolog F 1,92 3,79 7,54E-04 (avian) 42 13670 204222_s_at GLIPR1 GLI pathogenesis-related 1 (glioma) 1,91 3,76 2,20E-04 65 11080 201631_s_at IER3 immediate early response 3 1,81 3,50 3,50E-06 101 36952 227697_at SOCS3 suppressor of cytokine signaling 3 1,76 3,38 4,71E-05 16 15514 206067_s_at WT1 Wilms tumor 1 1,74 3,34 1,87E-04 171 47873 238623_at NA NA 1,72 3,30 1,10E-04 600 14687 205239_at AREG amphiregulin (schwannoma-derived growth factor) 1,71 3,26 1,51E-03 256 36997 227742_at CLIC6 chloride intracellular channel 6 1,69 3,23 3,52E-04 14 15038 205590_at RASGRP1 RAS guanyl releasing protein 1 (calcium and DAG-regulated) 1,68 3,20 1,87E-04 55 33237 223961_s_at CISH cytokine inducible SH2-containing protein 1,67 3,19 6,49E-07 78 32152 222872_x_at OBFC2A oligonucleotide/oligosaccharide-binding fold containing 2A 1,66 3,15 1,23E-05 1969 32201 222921_s_at HEY2 hairy/enhancer-of-split related with YRPW motif 2 1,64 3,12 1,78E-02 122 13463 204015_s_at DUSP4 dual specificity phosphatase 4 1,61 3,06 5,97E-05 173 36466 227210_at NA NA 1,60 3,04 1,10E-04 117 40525 231270_at CA13 carbonic anhydrase XIII 1,59 3,02 5,62E-05 81 42339 233085_s_at OBFC2A oligonucleotide/oligosaccharide-binding