Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ammonium Formate As Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-Ααα-Methylbenzylamine
RSC Advances Ammonium Formate as Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-ααα-Methylbenzylamine Journal: RSC Advances Manuscript ID: RA-ART-01-2014-000462.R1 Article Type: Paper Date Submitted by the Author: 21-Feb-2014 Complete List of Authors: Miranda, Leandro S. M.; Federal University of Rio de Janeiro, Biocatalysis and Organic Synthesis Lab, Chemistry Institute de Souza, Rodrigo Octavio; Federal University of Rio de Janeiro, de Miranda, Amanda; Federal University of Rio de Janeiro, Page 1 of 21 RSC Advances Graphical Abstract RSC Advances Page 2 of 21 Ammonium Formate as Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-α- Methylbenzylamine Amanda S. de Miranda, [a] Rodrigo O. M. A. de Souza, [ a] Leandro S. M. Miranda [a]* Keywords: Dynamic kinetic resolution • racemic amines • continuous flow . ammonium formate. Abstract: Abstract: The chemoenzymatic dynamic kinetic resolution of (+/-)-α- Methylbenzylamine under continuous flow conditions in the presence of Pd/BaSO 4 as racemization catalyst and ammonium formate as reductant is described. Under the conditions developed good conversions and excellent enantiomeric excess are reported Page 3 of 21 RSC Advances Introduction Recently, continuous processing and biocatalysis have been elected as key green engineering research areas for sustainable manufacturing 1a and it is clear that joint efforts between these areas can lead to great improvements on continuous manufacturing in agreement with green chemistry principles 1b,c . Optically pure amines are ubiquitously present in nature and active pharmaceutical ingredients (APIs). However, their synthesis still represents an ongoing synthetic challenge that can be inferred by the great amount of work and methodologies dealing with this issue in the literature. -

Deleterious Effects of Formic Acid Without Salt Additives on the HILIC Analysis of Basic Compounds
HPLC TN-1040 Deleterious Effects of Formic Acid without Salt Additives on the HILIC Analysis of Basic Compounds A. Carl Sanchez and Monika Kansal Phenomenex, Inc., Torrance, CA, USA Abstract Formic acid is an often-used mobile phase additive for of weak acids increase. The pKa shifts can be quite significant adjusting pH in reversed phase liquid chromatography (RPLC), in the high organic environment used for HILIC. For example, especially when using mass spectrometric (MS) detection. This weak bases with aqueous pKa less than ~4 typically will not be practice has been carried over to hydrophilic interaction liquid protonated in HILIC mobile phases when 0.1 v/v % formic acid chromatography (HILIC) separations. However, the mechanisms is used. The pKa of the base is decreased in HILIC mobile phase of action and the relative importance of buffer cation and while the pKa of the formic acid is increased. The increased pKa anion are much different in HILIC than RPLC. For this reason of formic acid leads to an increase in mobile phase pH. The buffer selection in HILIC mode requires consideration of buffer, combination of these opposing changes in pKa results in 0.1 v/v analyte and chromatographic sorbent chemical properties to % formic acid being too weak to protonate bases with pKa < ~4. make an appropriate choice. Proper choice of buffer can make Therefore, formic acid can provide acceptable chromatographic the difference between success and failure with HILIC. In this performance for weak bases with aqueous pKa < ~4. However, paper, the behavior of formic acid with and without the addition basic compounds with aqueous pKa greater than ~4 can be of various salts on the HILIC separation of basic analytes is protonated under HILIC conditions with formic acid. -
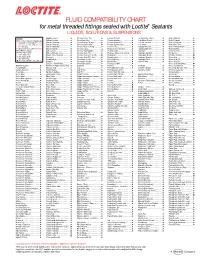
FLUID COMPATIBILITY CHART for Metal Threaded Fittings Sealed with Loctite¨ Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS
FLUID COMPATIBILITY CHART for metal threaded fittings sealed with Loctite® Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS LEGEND: Bagasse Fibers.......................... Chlorobenzene Dry ................... Ferrous Chloride ...................... Ion Exclusion Glycol ................. Nickel Chloride.......................... All Loctite® Anaerobic Sealants are Barium Acetate ........................ Chloroform Dry......................... Ferrous Oxalate......................... Irish Moss Slurry...................... Nickel Cyanide ......................... Compatible Including #242®, 243, Barium Carbonate..................... Chloroformate Methyl............... Ferrous Sulfate10%.................. Iron Ore Taconite ..................... Nickel Fluoborate ..................... 542, 545, 565, 567, 569, 571, 572, Barium Chloride........................ Chlorosulfonic Acid .................. Ferrous Sulfate (Sat)................. Iron Oxide ................................ Nickel Ore Fines ....................... 577, 580, 592 Barium Hydroxide..................... Chrome Acid Cleaning .............. Fertilizer Sol ............................. Isobutyl Alcohol ....................... Nickel Plating Bright ................. † Use Loctite® #270, 271™, 277, 554 Barium Sulfate.......................... Chrome Liquor.......................... Flotation Concentrates.............. Isobutyraldehyde ..................... Nickel Sulfate ........................... Not Recommended Battery Acid .............................. Chrome Plating -

Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017
EPA/600/R-17/356 | September 2017 www.epa.gov/homeland-security-research Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 Office of Research and Development Homeland Security Research Program This page left intentionally blank EPA/600/R-17/356 | September 2017 Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 UNITED STATES ENVIRONMENTAL PROTECTION AGENCY Cincinnati, OH 45268 Office of Research and Development Homeland Security Research Program Disclaimer Disclaimer The U.S. Environmental Protection Agency (EPA) through its Office of Research and Development funded and managed the research described here under Contract EP-C-15-012 to CSRA Inc. This document is undergoing review and has not been approved for publication. The contents reflect the views of the contributors and technical work groups and do not necessarily reflect the views of the Agency. Mention of trade names or commercial products in this document or in the methods referenced in this document does not constitute endorsement or recommendation for use. Questions concerning this document or its application should be addressed to: Romy Campisano National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King Drive Cincinnati, OH 45268 (513) 569-7016 [email protected] Kathy Hall National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Making LC Methods MS Friendly
Making LC Methods MS Friendly Mark Powell Applications Engineer Columns and Supplies Technical Support 8 October 2013 Topics •LC/MS ionization techniques •ESI •APCI •APPI •Appropriate conditions •Volatile buffers for MS •Ion pair chromatography •HILIC •Appropriate columns •Column diameter •Bonded phase •Particle size •Adapting existing methods to LC/MS •Maximizing Sensitivity •Minimize extra column volume •Avoiding interferences •Sample preparation LC/MS Techniques and Applications •Atmospheric pressure ionization (API) •Three typical API methods: •ESI - electrospray ionization •APCI - atmospheric pressure chemical ionization •APPI - atmospheric pressure photoionization •Appropriate ionization method depends largely on analyte polarity •Positive ion mode (protonation) or negative ion mode (deprotonation) •Masses measured as mass to charge ratio (m/z) Applicability of Atmospheric Pressure Ionization Techniques Electrospray Ionization • Most common ionization technique • Used for high and low molecular weight compounds • Ions are formed in solution and then the droplets are evaporated • Analyte volatility not required • Compounds containing heteroatoms such as N, S, and O typically analyze well • Can form multiply-charged ions • Like UV detection, ESI is concentration sensitive • ESI is generally more sensitive for samples that are ionized in solution Electrospray Ionization Electrospray Ions Nebulizer (gas Heated nitrogen drying gas shown in red) Solvent spray + + Dielectric capillary entrance APCI and APPI Sources •APCI •Analyte and mobile -

Ammonium Formate Afm
AMMONIUM FORMATE AFM CAUTIONARY RESPONSE INFORMATION 4. FIRE HAZARDS 7. SHIPPING INFORMATION 4.1 Flash Point: 7.1 Grades of Purity: Analytical grade; Organic Common Synonyms Solid White Weak ammonia Not flammable chemical grade odor Formic acid, ammonium salt 4.2 Flammable Limits in Air: Not pertinent 7.2 Storage Temperature: Ambient 4.3 Fire Extinguishing Agents: Water, foam 7.3 Inert Atmosphere: No requirement Sinks and mixes slowly with water. 4.4 Fire Extinguishing Agents Not to Be 7.4 Venting: Open Used: Currently not available 7.5 IMO Pollution Category: Currently not available Stop discharge if possible. Keep people away. 4.5 Special Hazards of Combustion Avoid contact with solid and dust; avoid inhalation. Products: Toxic and irritating ammonia 7.6 Ship Type: Currently not available Isolate and remove discharged material. and formic acid gases may form in fire. 7.7 Barge Hull Type: Currently not available Notify local health and pollution control agencies. 4.6 Behavior in Fire: Currently not available Protect water intakes. 4.7 Auto Ignition Temperature: Not pertinent 8. HAZARD CLASSIFICATIONS Combustible 4.8 Electrical Hazards: Not pertinent 8.1 49 CFR Category: Not listed Fire POISONOUS GASES MAY BE PRODUCED IN FIRE. 4.9 Burning Rate: Not pertinent 8.2 49 CFR Class: Not pertinent Irritating gases may be produced when heated. 4.10 Adiabatic Flame Temperature: Not 8.3 49 CFR Package Group: Not listed. Wear goggles, self-contained breathing apparatus and rubber overclothing pertinent (including gloves). 4.11 Stoichometric Air to Fuel Ratio: Not 8.4 Marine Pollutant: No Extinguish with water or foam. -

Lab & Reagent Chemicals Pat Impex
LAB CHEMICALS & REAGENTS – FINE CHEMICALS Manufacturer, Suppliers & Distributor MANUFACTURER SUPPLIER AND DISTRIBUTOR OF LABORATORY CHEMICALS, FINE CHEMICALS, AR LR GRADE, SUPERFINE CHEMICALS IN VADODARA, GUJARAT, INDIA. Our Introduction LAB CHEMICALS, REAGENTS GRADE CHEMICALS, BULK PACKING Our Range of Products • Laboratory chemicals & Reagents • Fine Chemicals AR LR ACS PH EUR • General Chemicals by Trading • Nanotechnology chemicals LABORATORY CHEMICALS, REAGENTS, FINE CHEMICALS • Acetamide Pure • Acetanilide Extrapure • Adipic Acid Pure • Acetanilide Exiplus • Aerosil 200 (Fumed Silica Gel) • Acetic Acid Glacial Extrapure • Agar Powder Regular Grade • Acetic Acid Glacial Extrapure Ar • Agar Powder Exiplus • Acetic Acid Glacial Extrapure Ar,acs,exiplus • Agar Powder Extrapure Bacto Grade • Acetic Acid Dried • Agar Granulated Bacto Grade • Acetic Acid For Hplc • Ajowan Seed Oil Extrapure • Acetic Acid Glacial For Molecular Biology • L-alanine Extrapure Chr • Acetone Pure • Aliquat 336 • Acetone Extrapure Ar • Alloxan Monohydrate • Acetone Extrapure Ar,acs,exiplus • Allyl Alcohol Pure • Acetone Gc-hs • Allylamine Pure • Acetone For Hplc & Uv Spectroscopy • Allyl Bromide Pure • Acetone For Uv Spectroscopy • Almond Oil Pure • Acetone Electronic Grade • Aluminium Ammonium Sulphate Dodecahydrate Pure • Acetone For Molecular Biology • Aluminium Ammonium Sulphate Dodecahydrate Extrapure Ar • Acetone -d6 For Nmr Spectroscopy • Aliminium Atomic Absorption Std.soln.aas • Acetonitrile(Acn)extrapure • Aluminium Metal Powder • Acetonitrile(Acn)extrapure -

Increase Analytical Accuracy LC/MS: Solvents, Blends, Standards, Surfactants
Increase Analytical Accuracy LC/MS: Solvents, Blends, Standards, Surfactants Reliability. Purity. Certainty. Discover. Synthesize. Analyze. Customize. Chemicals from Thermo Fisher Scientific brands offer a wide range of reagents, blends and standards designed to deliver more accurate results for liquid chromatography and mass spectrometry (LC/MS) analysis. The products in this brochure are specifically formulated, tested and validated on the latest instruments from leading brands to offer superior performance for LC/MS, and they’re conveniently provided in ready-to-use sizes with innovative packaging. Table of Contents Surfactants .........................................................1 Standards ...........................................................2 Specialty Flush Solutions and Probe Washes ......2 Mobile Phase Blends, Optima LC/MS Grade .......3 Solvents, Optima LC/MS Grade ..........................5 Reagents, Optima LC/MS Grade ........................6 Innovative Packaging .........................................10 Surfactants Alfa Aesar™ surfactants are compatible with mass spectrometry used in proteomics analysis. The compounds listed here showed minimal matrix interference in proteomics target analysis by mass spectrometry. Description Pack Size Cat. No. Anionic Acid Labile Surfactant I (AALS I) 5mg AAJ67647LB0 Anionic Acid Labile Surfactant II (AALS II) 5mg AAJ67832LB0 Cationic Acid Labile Surfactant I (CALS I) 5mg AAJ67743LB0 Cationic Acid Labile Surfactant II (CALS II) 5mg AAJ67525LB0 Non-Ionic Acid Labile Surfactant I (NALS I) 5mg AAJ67592LB0 Non-Ionic Acid Labile Surfactant II (NALS II) 5mg AAJ67682LB0 Zwitterionic Acid Labile Surfactant I (ZALS I) 5mg AAJ67729LB0 Zwitterionic Acid Labile Surfactant II (ZALS II) 5mg AAJ67721LB0 1 Discover. Synthesize. Analyze. Customize. Preserving Product Integrity Standards Standardization verifies instrument function and its suitability for the next analysis. By regularly calibrating your instruments using reliable chromatography standards, you’ll always have confidence in the accuracy of your data. -

UNION CHRISTIAN COLLEGE Aluva- 683 102 (Affiliated to Mahatma Gandhi University, Kottayam) Ernakulam Dt
Post Box: 5 UNION CHRISTIAN COLLEGE Aluva- 683 102 (Affiliated to Mahatma Gandhi University, Kottayam) Ernakulam Dt. Kerala Fax: 91-484- 2607534 Accredited by NAAC with “A” Grade, Cycle-4 91-484-2606033, 2609194 Email: [email protected] LIMITED TENDER NOTICE No. Department: Chemistry Sealed tenders are invited for the supply of the following Items: CHEMICALS ACETANILIDE ACETONE ACETONE COMML ACETOPHENONE ACETIC ACID GLACIAL ACID HYDROCHLORIC PURE ACID HYDROCHLORIC COMML ACID NITRIC COMML ACID NITRIC PURE ACID ORTHO- PHOSPHORIC ACID SULPHURIC COMML ACID SULPHURIC PURE AGAR AGAR ALCOHOL AMYL ISO – AMYL ALCOHOL ALUMINA (NEUTRAL) ALUMINIUM CHLORIDE ALUMINIUM NITRATE ALUMINIUM SULPHATE AMINO NAPHTHALENE- ALPHA AMINO PHENOL AMMONIA SOLUTION ER / AR AMMONIUM CARBONATE AMMONIUM ACETATE AMMONIUM BROMIDE AMMONIUM BORATE AMMONIUM CERIC SULPHATE AMMONIUM CERIC NITRATE AMMONIUM CHLORIDE AR AMMONIUM CHLORIDE PURE AMMONIUM CHLORIDE COMML AMMONIUM FORMATE AMMONIUM FLURIDE AMMONIUM NITRITE AMMONIUM NITRATE AMMONIUM NICKEL(II) SULPHATE AMMONIUM OXALATE AMMONIUM PURPURATE (MUREXIDE) AMMONIUM SULPHATE AMMONIUM SULPHIDE AMMONIUM IRON SULPHATE AMMONIUM FERROUS SULFATE AMMONIUM MOLYBDATE AMMONIUM IRON (II) SULPHATE 2-AMINO PYRIDINE Mohr's Salt AMMONIUM THIO CYNATE AMMONIUM THIOSULPHATE ANILINE ANISOLE PARA ANISALDEHYDE ANTHRACENE ANTHRANILIC ACID BARIUM CHLORIDE AR BARIUM CHLORIDE PURE BARIUM NITRATE BENZALDEHYDE BENZENE BENZOIC ACID BENZOIN BENZOPHENONE BENZIL BENZYL ALCOHOL BROMINE BIPHENYLE CALCIUM CHLORIDE (ANHYDROUS) CALCIUM FLURIDE CALCIUM HYDROXIDE CALCIUM -

Chemistry of Strontium in Natural Water
Chemistry of Strontium in Natural Water GEOLOGICAL SURVEY WATER-SUPPLY PAPER 1496 This water-supply paper was printed as separate chapters A-D UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1963 UNITED STATES DEPARTMENT OF THE INTERIOR STEWART L. UDALL, Secretary GEOLOGICAL SURVEY Thomas B. Nolan, Director The U.S. Geological Survey Library has cataloged this publication as follows: U.S. Geological Survey. Chemistry of strontium in natural water. Washington, U.S. Govt. Print. Off., 1962. iii, 97 p. illus., diagrs., tables. 24 cm. (Its Water-supply paper 1496) Issued as separate chapters A-D. Includes bibliographies. 1. Strontium. 2. Water-Analysis. I. Title. (Series) CONTENTS [The letters in parentheses preceding the titles are those used to designate the separate chapters] Page (A) A survey of analytical methods for the determination of strontium in natural water, by C. Albert Horr____________________________ 1 (B) Copper-spark method for spectrochemical determination of strontirm in water, by Marvin W. Skougstad-______-_-_-_--_~__-___-_- 19 (C) Flame photometric determination of strontium in natural water, by C. Albert Horr_____._____._______________... 33 (D) Occurrence and distribution of strontium in natural water, by Margin W. Skougstad and C. Albert Horr____________.___-._-___-. 55 iii A Survey of Analytical Methods fc r The Determination of Strontium in Natural Water By C. ALBERT HORR CHEMISTRY OF STRONTIUM IN NATURAL rVATER GEOLOGICAL SURVEY WATER-SUPPLY PAPER 1496-A This report concerns work done on behalf of the U.S. Atomic Energy Commission and is published with the permission of the Commission UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1959 UNITED STATES DEPARTMENT OF THE INTERIOR FRED A. -

Chemical Compatibility Storage Group
CHEMICAL SEGREGATION Chemicals are to be segregated into 11 different categories depending on the compatibility of that chemical with other chemicals The Storage Groups are as follows: Group A – Compatible Organic Acids Group B – Compatible Pyrophoric & Water Reactive Materials Group C – Compatible Inorganic Bases Group D – Compatible Organic Acids Group E – Compatible Oxidizers including Peroxides Group F– Compatible Inorganic Acids not including Oxidizers or Combustible Group G – Not Intrinsically Reactive or Flammable or Combustible Group J* – Poison Compressed Gases Group K* – Compatible Explosive or other highly Unstable Material Group L – Non-Reactive Flammable and Combustible, including solvents Group X* – Incompatible with ALL other storage groups The following is a list of chemicals and their compatibility storage codes. This is not a complete list of chemicals, but is provided to give examples of each storage group: Storage Group A 94‐75‐7 2,4‐D (2,4‐Dichlorophenoxyacetic acid) 94‐82‐6 2,4‐DB 609-99-4 3,5-Dinitrosalicylic acid 64‐19‐7 Acetic acid (Flammable liquid @ 102°F avoid alcohols, Amines, ox agents see SDS) 631-61-8 Acetic acid, Ammonium salt (Ammonium acetate) 108-24-7 Acetic anhydride (Flammable liquid @102°F avoid alcohols see SDS) 79‐10‐7 Acrylic acid Peroxide Former 65‐85‐0 Benzoic acid 98‐07‐7 Benzotrichloride 98‐88‐4 Benzoyl chloride 107-92-6 Butyric Acid 115‐28‐6 Chlorendic acid 79‐11‐8 Chloroacetic acid 627‐11‐2 Chloroethyl chloroformate 77‐92‐9 Citric acid 5949-29-1 Citric acid monohydrate 57-00-1 Creatine 20624-25-3