Practical Approach to Green Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ammonium Formate As Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-Ααα-Methylbenzylamine
RSC Advances Ammonium Formate as Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-ααα-Methylbenzylamine Journal: RSC Advances Manuscript ID: RA-ART-01-2014-000462.R1 Article Type: Paper Date Submitted by the Author: 21-Feb-2014 Complete List of Authors: Miranda, Leandro S. M.; Federal University of Rio de Janeiro, Biocatalysis and Organic Synthesis Lab, Chemistry Institute de Souza, Rodrigo Octavio; Federal University of Rio de Janeiro, de Miranda, Amanda; Federal University of Rio de Janeiro, Page 1 of 21 RSC Advances Graphical Abstract RSC Advances Page 2 of 21 Ammonium Formate as Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-α- Methylbenzylamine Amanda S. de Miranda, [a] Rodrigo O. M. A. de Souza, [ a] Leandro S. M. Miranda [a]* Keywords: Dynamic kinetic resolution • racemic amines • continuous flow . ammonium formate. Abstract: Abstract: The chemoenzymatic dynamic kinetic resolution of (+/-)-α- Methylbenzylamine under continuous flow conditions in the presence of Pd/BaSO 4 as racemization catalyst and ammonium formate as reductant is described. Under the conditions developed good conversions and excellent enantiomeric excess are reported Page 3 of 21 RSC Advances Introduction Recently, continuous processing and biocatalysis have been elected as key green engineering research areas for sustainable manufacturing 1a and it is clear that joint efforts between these areas can lead to great improvements on continuous manufacturing in agreement with green chemistry principles 1b,c . Optically pure amines are ubiquitously present in nature and active pharmaceutical ingredients (APIs). However, their synthesis still represents an ongoing synthetic challenge that can be inferred by the great amount of work and methodologies dealing with this issue in the literature. -

Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives
Article Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives Tomislav Portada, Davor Margetić and Vjekoslav Štrukil * Division of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička cesta 54, 10000 Zagreb, Croatia; [email protected] (T.P.); [email protected] (D.M.) * Correspondence: [email protected]; Tel.: +385‐1‐468‐0197 Received: 15 November 2018; Accepted: 29 November 2018; Published: date Abstract: Mechanochemical ball milling catalytic transfer hydrogenation (CTH) of aromatic nitro compounds using readily available and cheap ammonium formate as the hydrogen source is demonstrated as a simple, facile and clean approach for the synthesis of substituted anilines and selected pharmaceutically relevant compounds. The scope of mechanochemical CTH is broad, as the reduction conditions tolerate various functionalities, for example nitro, amino, hydroxy, carbonyl, amide, urea, amino acid and heterocyclic. The presented methodology was also successfully integrated with other types of chemical reactions previously carried out mechanochemically, such as amide bond formation by coupling amines with acyl chlorides or anhydrides and click‐type coupling reactions between amines and iso(thio)cyanates. In this way, we showed that active pharmaceutical ingredients Procainamide and Paracetamol could be synthesized from the respective nitro‐precursors on milligram and gram scale in excellent isolated yields. Keywords: mechanochemistry; catalytic transfer hydrogenation; aromatic nitro derivatives; ammonium formate; aging; ball milling; synthesis 1. Introduction Catalytic hydrogenation is one of the most significant functional group transformation reactions in organic synthesis and numerous procedures and reagents have been developed for that purpose [1,2]. As such, the hydrogenation reaction plays one of the key roles in many industrially important processes, for example hydrogenation of carbon monoxide to methanol or in food industry for the conversion of unsaturated vegetable oils into saturated triglycerides [3]. -

Chemistry Inventory; Fall
CHEMISTRY FALL 2005 MSDS Mfg.'s Name Chemical Name Quantity Stored Storage Conditions (on file = 9) Aluminum 9 1.5 kg Aluminum chloride, anhydrous, 98.5% 9 0.2 kg Aluminum chloride · 6H2O 9 0.5 kg Aluminum hydroxide 9 0.5 kg Aluminum nitrate 9 0.5 kg Aluminum sulfate 9 0.5 kg Ammonia, concentrated 9 4.0 L Ammonium acetate 9 0.2 kg Ammonium chloride 9 Ammonium dihydrogen phosphate (monobasic) 9 0.4 kg J.T. Baker Ammonium hydrogen phosphate (dibasic) No 0.5 kg Ammonium nitrate 9 2.5 kg Ammonium oxalate 9 0.7 kg Ammonium peroxydisulfate 9 0.5 kg Ammonium sulfate 9 0.2 kg Antimony 9 0.4 kg Barium chloride, anhydrous 9 2.5 kg Barium chloride · 2H2O 9 2.5 kg Barium nitrate 9 0.8 kg Bismuth 9 2.0 kg Boric Acid 9 0.4 kg Brass 9 Bromine 9 2.5 kg Cadmium 9 0.1 kg Cadmium nitrate 9 0.3 kg Calcium acetate · xH2O 9 0.5 kg Calcium carbide 9 1.0 kg Calcium carbonate 9 2.2 kg Calcium chloride 9 1.0 kg Calcium hydroxide 9 0.3 kg Calcium nitrate · 4H2O 9 1.0 kg Calcium oxide 9 0.3 kg Calcium sulfate · 2H2O 9 1.0 kg Carbon 9 0.1 kg Ceric ammonium nitrate 9 0.5 kg Cesium chloride 9 0.01 kg Chromium 9 0.01 kg Chromium chloride 9 0.5 kg Chromium nitrate 9 0.5 kg Cobalt 9 0.025 kg Cobalt chloride 9 0.7 kg Cobalt nitrate 9 0.6 kg Copper (assorted) 9 4.0 kg Copper acetate 9 0.05 kg Copper chloride 9 0.1 kg Copper nitrate 9 3.5 kg Copper oxide 9 0.4 kg Cupric sulfate, anhydrous 9 0.5 kg Cupric sulfate · 5H2O 9 2.75 kg EDTA 9 0.6 kg Iodine 9 2.0 kg Iron (assorted) 9 5.0 kg MSDS Mfg.'s Name Chemical Name Quantity Stored Storage Conditions (on file = 9) Ferric ammonium -

Deleterious Effects of Formic Acid Without Salt Additives on the HILIC Analysis of Basic Compounds
HPLC TN-1040 Deleterious Effects of Formic Acid without Salt Additives on the HILIC Analysis of Basic Compounds A. Carl Sanchez and Monika Kansal Phenomenex, Inc., Torrance, CA, USA Abstract Formic acid is an often-used mobile phase additive for of weak acids increase. The pKa shifts can be quite significant adjusting pH in reversed phase liquid chromatography (RPLC), in the high organic environment used for HILIC. For example, especially when using mass spectrometric (MS) detection. This weak bases with aqueous pKa less than ~4 typically will not be practice has been carried over to hydrophilic interaction liquid protonated in HILIC mobile phases when 0.1 v/v % formic acid chromatography (HILIC) separations. However, the mechanisms is used. The pKa of the base is decreased in HILIC mobile phase of action and the relative importance of buffer cation and while the pKa of the formic acid is increased. The increased pKa anion are much different in HILIC than RPLC. For this reason of formic acid leads to an increase in mobile phase pH. The buffer selection in HILIC mode requires consideration of buffer, combination of these opposing changes in pKa results in 0.1 v/v analyte and chromatographic sorbent chemical properties to % formic acid being too weak to protonate bases with pKa < ~4. make an appropriate choice. Proper choice of buffer can make Therefore, formic acid can provide acceptable chromatographic the difference between success and failure with HILIC. In this performance for weak bases with aqueous pKa < ~4. However, paper, the behavior of formic acid with and without the addition basic compounds with aqueous pKa greater than ~4 can be of various salts on the HILIC separation of basic analytes is protonated under HILIC conditions with formic acid. -

Step-By-Step Guide to Better Laboratory Management Practices
Step-by-Step Guide to Better Laboratory Management Practices Prepared by The Washington State Department of Ecology Hazardous Waste and Toxics Reduction Program Publication No. 97- 431 Revised January 2003 Printed on recycled paper For additional copies of this document, contact: Department of Ecology Publications Distribution Center PO Box 47600 Olympia, WA 98504-7600 (360) 407-7472 or 1 (800) 633-7585 or contact your regional office: Department of Ecology’s Regional Offices (425) 649-7000 (509) 575-2490 (509) 329-3400 (360) 407-6300 The Department of Ecology is an equal opportunity agency and does not discriminate on the basis of race, creed, color, disability, age, religion, national origin, sex, marital status, disabled veteran’s status, Vietnam Era veteran’s status or sexual orientation. If you have special accommodation needs, or require this document in an alternate format, contact the Hazardous Waste and Toxics Reduction Program at (360)407-6700 (voice) or 711 or (800) 833-6388 (TTY). Table of Contents Introduction ....................................................................................................................................iii Section 1 Laboratory Hazardous Waste Management ...........................................................1 Designating Dangerous Waste................................................................................................1 Counting Wastes .......................................................................................................................8 Treatment by Generator...........................................................................................................12 -

The Application of Controlled Radical Polymerization Processes on the Graft Copolymerization of Hydrophobic Substituents Onto Guar Gum and Guar Gum Derivatives
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2007 The pplica ation of controlled radical polymerization processes on the graft copolymerization of hydrophobic substituents onto guar gum and guar gum derivatives Veronica Holmes Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Chemistry Commons Recommended Citation Holmes, Veronica, "The ppa lication of controlled radical polymerization processes on the graft opoc lymerization of hydrophobic substituents onto guar gum and guar gum derivatives" (2007). LSU Doctoral Dissertations. 3220. https://digitalcommons.lsu.edu/gradschool_dissertations/3220 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. THE APPLICATION OF CONTROLLED RADICAL POLYMERIZATION PROCESSES ON THE GRAFT COPOLYMERIZATION OF HYDROPHOBIC SUBSTITUENTS ONTO GUAR GUM AND GUAR GUM DERIVATIVES A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College In partial fulfillment of the Requirements for the degree of Doctor of Philosophy In The Department of Chemistry By Veronica Holmes B.S., Southern University A&M, Baton Rouge, LA, 1999 May, 2007 Acknowledgements My matriculation through graduate school would not have been possible without the support and guidance of Dr. William H. Daly. His kindness and patience during my graduate endeavors has been unprecedented. Along with the support of past and present graduate students in our research lab, I have had an excellent support system for the duration of time I have spent with this research. -

Ceric Ammonium Nitrate (CAN) Catalyzed Baeyer-Villiger Oxidation of Carbonyl Compounds, Specially 20-Oxosteroids
Indian Journal of Chemistry Vol. 43B, June 2004, pp . 1275-1281 Ceric ammonium nitrate (CAN) catalyzed Baeyer-Villiger oxidation of carbonyl compounds, specially 20-oxosteroids Papori Goswami, Saroj Hazarika, Archana M Das & Pritish Chowdhury* Natural Products Chemistry Division, Regional Research Laboratory, Jorhat 785006, India e-mail: [email protected] Received 4 February 2003; accepted (revised) 10 December 2003 The role of ceric ammonium nitrate (CAN) as an effective catalyst in the peracid induced Baeyer-Villiger oxidation of carbonyl compounds with special reference to steroids has been demonstrated. IPC: Int.C1.7 C 07 K 1/00 Ceric ammonium nitrate (CAN) finds application in temperature even when kept for more than 48 hr. Fur synthetic organic chemistry for various chemical ther, GLC experiment confirmed 40-55% conversion 2 transformations, viz., nitration 1, nitroacetamidation , of benzophenone 12 to phenyl benzoate 12a in 5 hr complex formation with various alcohols3 etc. Its role when CAN was used as a catalyst along with peracid as single electron oxidant has been reported in a num whereas only 8% conversion was observed in the ab 4 8 ber of publications including some recent reviews - . sence of CAN when kept for more than 24 hr. During CAN-induced oxidative radical transformations of our investigation, we also found that for all the cases 9 steroids have also been reported . We too have re (substrates 5-8, 10, 11, 13-15) the oxidation furni shed ported lO the catalytic action of CAN in the esterifica only one isomer viz. 5a-8a, lOa, lla, 13a-15a as con tion of carboxylic acids in very high yield . -

Quinone Formation Via Ceric Ammonium Nitrate Oxidations of 2-Alkyl-1,4-Dialkoxybenzenes
Quinone Formation via Ceric Ammonium Nitrate Oxidations of 2-Alkyl-1,4-dialkoxybenzenes by Alexander Linwood Simmons April, 2016 Director of Thesis: Brian E. Love, PhD Major Department: Chemistry Quinones are cyclohexadiendiones that have a variety of uses ranging from medical applications to synthetic building blocks.1 Medicinal applications stem from the potent biological activity (e.g. antitumor and antibiotic) these compounds and some derivatives possess.2, 3 The most common preparation method to access these compounds is oxidative demethylation of hydroquinone dimethyl ethers (1, R1=R3: Me) typically using ceric ammonium nitrate (CAN) as seen in Figure 1. Oxidation using CAN can yield a product mixture of the (mono)quinone (2) and the symmetric dimeric quinone (3). Previous work4, 5 in our group has resulted in the development of several protocols for altering the monoquinone to diquinone ratio by altering reaction conditions (e.g. substrate concentration, mode of addition, etc.). The current focus further explores manipulation of this ratio and reaction efficacy through substrate solubility and cerium coordination. We will discuss how ether linkages of various hydrophobicities and coordination modes change product outcome and if altering a single ether linkage (R1) or both linkages (both R1 and R3) affect the product ratio. 1 2 3 Figure 1 – General Substrate Reaction Quinone Formation via Ceric Ammonium Nitrate Oxidations of 2-Alkyl-1,4-dialkoxybenzenes A Thesis Presented To the Faculty of the Department of Chemistry East Carolina University In Partial Fulfillment of the Requirements for the Degree Master of Science in Chemistry by Alexander Linwood Simmons April, 2016 © Alexander Linwood Simmons, 2016 Quinone Formation via Ceric Ammonium Nitrate Oxidations of 2-Alkyl-1,4-dialkoxybenzenes by Alexander Linwood Simmons Approved by: Director of Thesis: ____________________________________________________________ Brian E. -
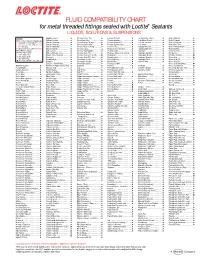
FLUID COMPATIBILITY CHART for Metal Threaded Fittings Sealed with Loctite¨ Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS
FLUID COMPATIBILITY CHART for metal threaded fittings sealed with Loctite® Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS LEGEND: Bagasse Fibers.......................... Chlorobenzene Dry ................... Ferrous Chloride ...................... Ion Exclusion Glycol ................. Nickel Chloride.......................... All Loctite® Anaerobic Sealants are Barium Acetate ........................ Chloroform Dry......................... Ferrous Oxalate......................... Irish Moss Slurry...................... Nickel Cyanide ......................... Compatible Including #242®, 243, Barium Carbonate..................... Chloroformate Methyl............... Ferrous Sulfate10%.................. Iron Ore Taconite ..................... Nickel Fluoborate ..................... 542, 545, 565, 567, 569, 571, 572, Barium Chloride........................ Chlorosulfonic Acid .................. Ferrous Sulfate (Sat)................. Iron Oxide ................................ Nickel Ore Fines ....................... 577, 580, 592 Barium Hydroxide..................... Chrome Acid Cleaning .............. Fertilizer Sol ............................. Isobutyl Alcohol ....................... Nickel Plating Bright ................. † Use Loctite® #270, 271™, 277, 554 Barium Sulfate.......................... Chrome Liquor.......................... Flotation Concentrates.............. Isobutyraldehyde ..................... Nickel Sulfate ........................... Not Recommended Battery Acid .............................. Chrome Plating -

Optimization of Ceric Ammonium Nitrate Initiated Graft Copolymerization of Acrylonitrile Onto Chitosan
Journal of Water Resource and Protection, 2011, 3, 380-386 doi:10.4236/jwarp.2011.36048 Published Online June 2011 (http://www.scirp.org/journal/jwarp) Optimization of Ceric Ammonium Nitrate Initiated Graft Copolymerization of Acrylonitrile onto Chitosan Arumugam Shanmugapriya1, Ramya Ramammurthy2, Venkatachalam Munusamy3 , Sudha N. Parapurath4 1Bharathiar University, Coimbatore, Tamil Nadu, India 2Department of Chemistry, Manonmaniam Sundaranar University, Tirunelveli, Tamil Nadu, India 3Department of Chemistry, Dravidian University, Kuppam, Andhra Pradesh, India 4DKM College, Thiruvalluvar University, Vellore, Tamil Nadu, India E-mail: [email protected] Received March 14, 2011; revised April 21, 2011; accepted May 25, 2011 Abstract In the present work, graft copolymerization of polyacrylonitrile onto chitosan has been carried out in the pre- sence of ceric ammonium nitrate redox initiator. Optimization of grafting of polyacrylonitrile onto chitosan was performed by varying the process parameters such as ceric ammonium nitrate (CAN) concentration, polyacrylonitrile concentration and reaction time to study their influence on percent grafting and grafting ef- ficiency. The optimum reaction conditions obtained for grafting of acrylonitrile onto chitosan were reaction time 55 mins, CAN concentration 1% in Con. HNO3, and polyacrylonitrile concentration 0.75 mol/L. The characterization of the grafted products by means of FTIR, thermal analysis, X-ray diffraction and scanning electron microscopy furnished the evidence of grafting of polyacrylonitrile onto chitosan. Keywords: Chitosan-g-polyacrylonitrile, Ceric ammonium nitrate, Grafting efficiency, Grafting yield 1. Introduction synthetic polymers increased its properties tremendously. Besides these applications chitosan has few drawbacks Chitosan is a unique basic polysaccharide and partially such as acidic solubility, low thermal and mechanical deacetylated polymer of glucosamine obtained after alka- stability. -

Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017
EPA/600/R-17/356 | September 2017 www.epa.gov/homeland-security-research Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 Office of Research and Development Homeland Security Research Program This page left intentionally blank EPA/600/R-17/356 | September 2017 Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 UNITED STATES ENVIRONMENTAL PROTECTION AGENCY Cincinnati, OH 45268 Office of Research and Development Homeland Security Research Program Disclaimer Disclaimer The U.S. Environmental Protection Agency (EPA) through its Office of Research and Development funded and managed the research described here under Contract EP-C-15-012 to CSRA Inc. This document is undergoing review and has not been approved for publication. The contents reflect the views of the contributors and technical work groups and do not necessarily reflect the views of the Agency. Mention of trade names or commercial products in this document or in the methods referenced in this document does not constitute endorsement or recommendation for use. Questions concerning this document or its application should be addressed to: Romy Campisano National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King Drive Cincinnati, OH 45268 (513) 569-7016 [email protected] Kathy Hall National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King -

Ammonium Cerium (IV) Nitrate
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 4.3 Revision Date 10/03/2012 Print Date 02/20/2014 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Ammonium cerium(IV) nitrate Product Number : C3654 Brand : Sigma-Aldrich Supplier : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # (For : (314) 776-6555 both supplier and manufacturer) Preparation Information : Sigma-Aldrich Corporation Product Safety - Americas Region 1-800-521-8956 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Oxidizer, Toxic by ingestion, Irritant GHS Classification Oxidizing solids (Category 2) Acute toxicity, Oral (Category 4) Skin irritation (Category 2) Eye irritation (Category 2A) Specific target organ toxicity - single exposure (Category 3) GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H272 May intensify fire; oxidiser. H302 Harmful if swallowed. H315 Causes skin irritation. H319 Causes serious eye irritation. H335 May cause respiratory irritation. Precautionary statement(s) P220 Keep/Store away from clothing/ combustible materials. P261 Avoid breathing dust/ fume/ gas/ mist/ vapours/ spray. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. HMIS Classification Health hazard: 2 Flammability: 0 Physical hazards: 2 Sigma-Aldrich - C3654 Page 1 of 7 NFPA Rating Health hazard: 2 Fire: 0 Reactivity Hazard: 2 Special hazard.: OX Potential Health Effects Inhalation May be harmful if inhaled. Causes respiratory tract irritation. Skin May be harmful if absorbed through skin. Causes skin irritation. Eyes Causes eye irritation.