Quinone Formation Via Ceric Ammonium Nitrate Oxidations of 2-Alkyl-1,4-Dialkoxybenzenes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry Inventory; Fall
CHEMISTRY FALL 2005 MSDS Mfg.'s Name Chemical Name Quantity Stored Storage Conditions (on file = 9) Aluminum 9 1.5 kg Aluminum chloride, anhydrous, 98.5% 9 0.2 kg Aluminum chloride · 6H2O 9 0.5 kg Aluminum hydroxide 9 0.5 kg Aluminum nitrate 9 0.5 kg Aluminum sulfate 9 0.5 kg Ammonia, concentrated 9 4.0 L Ammonium acetate 9 0.2 kg Ammonium chloride 9 Ammonium dihydrogen phosphate (monobasic) 9 0.4 kg J.T. Baker Ammonium hydrogen phosphate (dibasic) No 0.5 kg Ammonium nitrate 9 2.5 kg Ammonium oxalate 9 0.7 kg Ammonium peroxydisulfate 9 0.5 kg Ammonium sulfate 9 0.2 kg Antimony 9 0.4 kg Barium chloride, anhydrous 9 2.5 kg Barium chloride · 2H2O 9 2.5 kg Barium nitrate 9 0.8 kg Bismuth 9 2.0 kg Boric Acid 9 0.4 kg Brass 9 Bromine 9 2.5 kg Cadmium 9 0.1 kg Cadmium nitrate 9 0.3 kg Calcium acetate · xH2O 9 0.5 kg Calcium carbide 9 1.0 kg Calcium carbonate 9 2.2 kg Calcium chloride 9 1.0 kg Calcium hydroxide 9 0.3 kg Calcium nitrate · 4H2O 9 1.0 kg Calcium oxide 9 0.3 kg Calcium sulfate · 2H2O 9 1.0 kg Carbon 9 0.1 kg Ceric ammonium nitrate 9 0.5 kg Cesium chloride 9 0.01 kg Chromium 9 0.01 kg Chromium chloride 9 0.5 kg Chromium nitrate 9 0.5 kg Cobalt 9 0.025 kg Cobalt chloride 9 0.7 kg Cobalt nitrate 9 0.6 kg Copper (assorted) 9 4.0 kg Copper acetate 9 0.05 kg Copper chloride 9 0.1 kg Copper nitrate 9 3.5 kg Copper oxide 9 0.4 kg Cupric sulfate, anhydrous 9 0.5 kg Cupric sulfate · 5H2O 9 2.75 kg EDTA 9 0.6 kg Iodine 9 2.0 kg Iron (assorted) 9 5.0 kg MSDS Mfg.'s Name Chemical Name Quantity Stored Storage Conditions (on file = 9) Ferric ammonium -

Step-By-Step Guide to Better Laboratory Management Practices
Step-by-Step Guide to Better Laboratory Management Practices Prepared by The Washington State Department of Ecology Hazardous Waste and Toxics Reduction Program Publication No. 97- 431 Revised January 2003 Printed on recycled paper For additional copies of this document, contact: Department of Ecology Publications Distribution Center PO Box 47600 Olympia, WA 98504-7600 (360) 407-7472 or 1 (800) 633-7585 or contact your regional office: Department of Ecology’s Regional Offices (425) 649-7000 (509) 575-2490 (509) 329-3400 (360) 407-6300 The Department of Ecology is an equal opportunity agency and does not discriminate on the basis of race, creed, color, disability, age, religion, national origin, sex, marital status, disabled veteran’s status, Vietnam Era veteran’s status or sexual orientation. If you have special accommodation needs, or require this document in an alternate format, contact the Hazardous Waste and Toxics Reduction Program at (360)407-6700 (voice) or 711 or (800) 833-6388 (TTY). Table of Contents Introduction ....................................................................................................................................iii Section 1 Laboratory Hazardous Waste Management ...........................................................1 Designating Dangerous Waste................................................................................................1 Counting Wastes .......................................................................................................................8 Treatment by Generator...........................................................................................................12 -

The Application of Controlled Radical Polymerization Processes on the Graft Copolymerization of Hydrophobic Substituents Onto Guar Gum and Guar Gum Derivatives
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 2007 The pplica ation of controlled radical polymerization processes on the graft copolymerization of hydrophobic substituents onto guar gum and guar gum derivatives Veronica Holmes Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Chemistry Commons Recommended Citation Holmes, Veronica, "The ppa lication of controlled radical polymerization processes on the graft opoc lymerization of hydrophobic substituents onto guar gum and guar gum derivatives" (2007). LSU Doctoral Dissertations. 3220. https://digitalcommons.lsu.edu/gradschool_dissertations/3220 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. THE APPLICATION OF CONTROLLED RADICAL POLYMERIZATION PROCESSES ON THE GRAFT COPOLYMERIZATION OF HYDROPHOBIC SUBSTITUENTS ONTO GUAR GUM AND GUAR GUM DERIVATIVES A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College In partial fulfillment of the Requirements for the degree of Doctor of Philosophy In The Department of Chemistry By Veronica Holmes B.S., Southern University A&M, Baton Rouge, LA, 1999 May, 2007 Acknowledgements My matriculation through graduate school would not have been possible without the support and guidance of Dr. William H. Daly. His kindness and patience during my graduate endeavors has been unprecedented. Along with the support of past and present graduate students in our research lab, I have had an excellent support system for the duration of time I have spent with this research. -

Ceric Ammonium Nitrate (CAN) Catalyzed Baeyer-Villiger Oxidation of Carbonyl Compounds, Specially 20-Oxosteroids
Indian Journal of Chemistry Vol. 43B, June 2004, pp . 1275-1281 Ceric ammonium nitrate (CAN) catalyzed Baeyer-Villiger oxidation of carbonyl compounds, specially 20-oxosteroids Papori Goswami, Saroj Hazarika, Archana M Das & Pritish Chowdhury* Natural Products Chemistry Division, Regional Research Laboratory, Jorhat 785006, India e-mail: [email protected] Received 4 February 2003; accepted (revised) 10 December 2003 The role of ceric ammonium nitrate (CAN) as an effective catalyst in the peracid induced Baeyer-Villiger oxidation of carbonyl compounds with special reference to steroids has been demonstrated. IPC: Int.C1.7 C 07 K 1/00 Ceric ammonium nitrate (CAN) finds application in temperature even when kept for more than 48 hr. Fur synthetic organic chemistry for various chemical ther, GLC experiment confirmed 40-55% conversion 2 transformations, viz., nitration 1, nitroacetamidation , of benzophenone 12 to phenyl benzoate 12a in 5 hr complex formation with various alcohols3 etc. Its role when CAN was used as a catalyst along with peracid as single electron oxidant has been reported in a num whereas only 8% conversion was observed in the ab 4 8 ber of publications including some recent reviews - . sence of CAN when kept for more than 24 hr. During CAN-induced oxidative radical transformations of our investigation, we also found that for all the cases 9 steroids have also been reported . We too have re (substrates 5-8, 10, 11, 13-15) the oxidation furni shed ported lO the catalytic action of CAN in the esterifica only one isomer viz. 5a-8a, lOa, lla, 13a-15a as con tion of carboxylic acids in very high yield . -

Optimization of Ceric Ammonium Nitrate Initiated Graft Copolymerization of Acrylonitrile Onto Chitosan
Journal of Water Resource and Protection, 2011, 3, 380-386 doi:10.4236/jwarp.2011.36048 Published Online June 2011 (http://www.scirp.org/journal/jwarp) Optimization of Ceric Ammonium Nitrate Initiated Graft Copolymerization of Acrylonitrile onto Chitosan Arumugam Shanmugapriya1, Ramya Ramammurthy2, Venkatachalam Munusamy3 , Sudha N. Parapurath4 1Bharathiar University, Coimbatore, Tamil Nadu, India 2Department of Chemistry, Manonmaniam Sundaranar University, Tirunelveli, Tamil Nadu, India 3Department of Chemistry, Dravidian University, Kuppam, Andhra Pradesh, India 4DKM College, Thiruvalluvar University, Vellore, Tamil Nadu, India E-mail: [email protected] Received March 14, 2011; revised April 21, 2011; accepted May 25, 2011 Abstract In the present work, graft copolymerization of polyacrylonitrile onto chitosan has been carried out in the pre- sence of ceric ammonium nitrate redox initiator. Optimization of grafting of polyacrylonitrile onto chitosan was performed by varying the process parameters such as ceric ammonium nitrate (CAN) concentration, polyacrylonitrile concentration and reaction time to study their influence on percent grafting and grafting ef- ficiency. The optimum reaction conditions obtained for grafting of acrylonitrile onto chitosan were reaction time 55 mins, CAN concentration 1% in Con. HNO3, and polyacrylonitrile concentration 0.75 mol/L. The characterization of the grafted products by means of FTIR, thermal analysis, X-ray diffraction and scanning electron microscopy furnished the evidence of grafting of polyacrylonitrile onto chitosan. Keywords: Chitosan-g-polyacrylonitrile, Ceric ammonium nitrate, Grafting efficiency, Grafting yield 1. Introduction synthetic polymers increased its properties tremendously. Besides these applications chitosan has few drawbacks Chitosan is a unique basic polysaccharide and partially such as acidic solubility, low thermal and mechanical deacetylated polymer of glucosamine obtained after alka- stability. -

Ammonium Cerium (IV) Nitrate
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 4.3 Revision Date 10/03/2012 Print Date 02/20/2014 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Ammonium cerium(IV) nitrate Product Number : C3654 Brand : Sigma-Aldrich Supplier : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # (For : (314) 776-6555 both supplier and manufacturer) Preparation Information : Sigma-Aldrich Corporation Product Safety - Americas Region 1-800-521-8956 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Oxidizer, Toxic by ingestion, Irritant GHS Classification Oxidizing solids (Category 2) Acute toxicity, Oral (Category 4) Skin irritation (Category 2) Eye irritation (Category 2A) Specific target organ toxicity - single exposure (Category 3) GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H272 May intensify fire; oxidiser. H302 Harmful if swallowed. H315 Causes skin irritation. H319 Causes serious eye irritation. H335 May cause respiratory irritation. Precautionary statement(s) P220 Keep/Store away from clothing/ combustible materials. P261 Avoid breathing dust/ fume/ gas/ mist/ vapours/ spray. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. HMIS Classification Health hazard: 2 Flammability: 0 Physical hazards: 2 Sigma-Aldrich - C3654 Page 1 of 7 NFPA Rating Health hazard: 2 Fire: 0 Reactivity Hazard: 2 Special hazard.: OX Potential Health Effects Inhalation May be harmful if inhaled. Causes respiratory tract irritation. Skin May be harmful if absorbed through skin. Causes skin irritation. Eyes Causes eye irritation. -
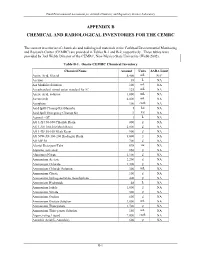
Appendix B Chemical and Radiological Inventories for the Cemrc
Final Environmental Assessment for Actinide Chemistry and Repository Science Laboratory APPENDIX B CHEMICAL AND RADIOLOGICAL INVENTORIES FOR THE CEMRC The current inventories of chemicals and radiological materials at the Carlsbad Environmental Monitoring and Research Center (CEMRC) are provided in Tables B-1 and B-2, respectively. These tables were provided by Joel Webb, Director of the CEMRC, New Mexico State University (Webb 2002). Table B-1. Onsite CEMRC Chemical Inventory Chemical Name Amount Units SARA Limit Acetic Acid, Glacial 5,400 mL NAa Acetone 38 L NA AA Modifier Solution 100 mL NA AccuStandard mixed anion standard for IC 125 mL NA Acetic Acid, solution 1,000 mL NA Acetonitrile 4,000 mL NA Acetylene 100 cu.ft. NA Acid Spill Cleanup Kit (Hazorb) 1 kit NA Acid Spill Emergency Cleanup Kit 3 kit NA Aerosol - OT 1 L NA AG 1-X4 50-100 Chloride Resin 800 g NA AG 1-X8 100-200 Mesh Resin 2,000 g NA AG 1-X8 50-100 Mesh Resin 900 g NA AG 50W-X8 100-200 Hydrogen Resin 3,000 g NA AG MP 50 700 g NA Alcojet Detergent/Tabs 698 oz NA Alumina, activated 850 g NA Aluminum Nitrate 2,100 g NA Ammonium Acetate 2,200 g NA Ammonium Chloride 1,300 g NA Ammonium Chloride Solution 300 mL NA Ammonium Citrate 100 g NA Ammonium hydrogenoxalate, hemihydrate 400 g NA Ammonium Hydroxide 48 L NA Ammonium Iodide 1,000 g NA Ammonium Nitrate 500 g NA Ammonium Oxalate 600 g NA Ammonium Oxalate Solution 2,000 mL NA Ammonium Thiocyanate 1,700 g NA Ammonium Thiocyanate Solution 150 mL NA Argon, refrig. -

Model Syllabus Chemistry Revised.Pdf
STATE MODEL SYLLABUS FOR UNDER GRADUATE COURSE IN CHEMISTRY (Bachelor of Science Examination) UNDER CHOICE BASED CREDIT SYSTEM Course structure of UG Chemistry Honours Semester Course Course Name Credits Total marks I AECC-I AECC-I 04 100 C-I Inorganic Chemistry-I 04 75 C-I Practical Inorganic Chemistry-I Lab 02 25 C-II Physical Chemistry-I 04 75 C-II Practical Physical Chemistry-I Lab 02 25 GE-I GE-I 04 75 GE-I Practical GE-I Lab 02 25 22 400 II AECC-II AECC-II 04 100 C-III Organic Chemistry-I 04 75 C-III Practical Organic Chemistry-I Lab 02 25 C-IV Physical Chemistry-II 04 75 C-IV Practical Physical Chemistry-II 02 25 GE-II GE-II 04 75 GE-II Practical GE-II Lab 02 25 22 400 III C-V Inorganic Chemistry-II 04 75 C-V Practical Inorganic Chemistry-II Lab 02 25 C-VI Organic Chemistry-II 04 75 C-VI Practical Organic Chemistry-II Lab 02 25 C-VII Physical Chemistry-III 04 75 C-VII Practical Physical Chemistry-III Lab 02 25 GE-III GE-III 04 75 GE-III Practical GE-III Lab 02 25 SECC-I SECC-I 04 100 28 500 IV C-VIII Inorganic Chemistry-III 04 75 C-VIII Practical Inorganic Chemistry-III Lab 02 25 C-IX Organic Chemistry-III 04 75 C-IX Practical Organic Chemistry-III Lab 02 25 C-X Physical Chemistry-IV 04 75 C-X Practical Physical Chemistry-IV Lab 02 25 GE-IV GE-IV (Theory) 04 75 GE-IV Practical GE-IV (Practical) 02 25 SECC-II SECC-II 04 100 28 500 V C-XI Organic Chemistry-IV 04 75 C-XI Practical Organic Chemistry-IV 02 25 C-XII Physical Chemistry-V 04 75 C-XII Practical Physical Chemistry-V 02 25 DSE-I DSE-I 04 75 DSE-I Practical DSE-I Lab 02 25 DSE-II DSE-II 04 75 DSE-II Practical DSE-II Lab 02 25 24 400 VI C-XIII Inorganic Chemistry- IV 04 75 C-XIII Practical Inorganic Chemistry-IV 02 25 C-XIV Organic Chemistry-V 04 75 C-XIV Practical Organic Chemistry-V 02 25 DSE-III DSE-III 04 75 DSE-III Practical DSE-III Lab 02 25 DSE-IV DSE-IV 04 75 DSE-IV Practical DSE-IV Lab 02 25 OR DSE-IV Dissertation 06 100* 24 400 TOTAL 148 2600 Discipline Specific Elective Papers: (Credit: 06 each) (4 papers to be selected by students of Chemistry Honours): DSE (1-IV) 1. -

Chemical Compatibility Storage Group
CHEMICAL SEGREGATION Chemicals are to be segregated into 11 different categories depending on the compatibility of that chemical with other chemicals The Storage Groups are as follows: Group A – Compatible Organic Acids Group B – Compatible Pyrophoric & Water Reactive Materials Group C – Compatible Inorganic Bases Group D – Compatible Organic Acids Group E – Compatible Oxidizers including Peroxides Group F– Compatible Inorganic Acids not including Oxidizers or Combustible Group G – Not Intrinsically Reactive or Flammable or Combustible Group J* – Poison Compressed Gases Group K* – Compatible Explosive or other highly Unstable Material Group L – Non-Reactive Flammable and Combustible, including solvents Group X* – Incompatible with ALL other storage groups The following is a list of chemicals and their compatibility storage codes. This is not a complete list of chemicals, but is provided to give examples of each storage group: Storage Group A 94‐75‐7 2,4‐D (2,4‐Dichlorophenoxyacetic acid) 94‐82‐6 2,4‐DB 609-99-4 3,5-Dinitrosalicylic acid 64‐19‐7 Acetic acid (Flammable liquid @ 102°F avoid alcohols, Amines, ox agents see SDS) 631-61-8 Acetic acid, Ammonium salt (Ammonium acetate) 108-24-7 Acetic anhydride (Flammable liquid @102°F avoid alcohols see SDS) 79‐10‐7 Acrylic acid Peroxide Former 65‐85‐0 Benzoic acid 98‐07‐7 Benzotrichloride 98‐88‐4 Benzoyl chloride 107-92-6 Butyric Acid 115‐28‐6 Chlorendic acid 79‐11‐8 Chloroacetic acid 627‐11‐2 Chloroethyl chloroformate 77‐92‐9 Citric acid 5949-29-1 Citric acid monohydrate 57-00-1 Creatine 20624-25-3 -

Analytical Reagents and Standards Pharmacopoeia Products
Analytical Reagents and Standards The Experts in Custom-made Standards - Organic & Inorganic Pharmacopoeia products www.cpachem.com Contact us at: mobile: (+359) 886 796501 Tel./fax: (+359) 42 607716 E-mail: [email protected] products Standards/Pharmacopoeia and Reagents Analytical [email protected] ISO Guide 34 | ISO 17025 | ISO 9001 is the world leader in Certified Reference Materials production (Custom and Stock) with both ISO Guide 34 and ISO/IEC 17025 accreditations. The four secrets of our success are: ✓ High-technology ✓ High-experienced staff ✓ High-quality ✓ High-speed Our scope covers: Inorganic Certified Reference Materials (CRMs) for AAS, ICP, ICP/MS and Ion Chromatography - Custom Inorganic CRMs - Single and Multi-components - Inorganic stock CRMs - Single and Multi-components - ICP and ICP/MS Internal standards - AAS and ICP Modifiers, Buffers and Reagents - IC Eluent concentrates 1 Organic Certified Reference Materials (CRMs) for GC and HPLC - Custom Organic CRMs - Single and Multi-components - CRMs acc. to ISO, EN, ASTM and EPA Methods - Contaminant CRMs - Single Component CRMs Analytical Certified Reference Materials (CRMs) and Reagents - Custom Analytical CRMs - Volumetric CRMs - Conductivity CRMs - pH Buffer CRMs -Reagents Our unique Computer-Aided- What makes us different? Manufacturing (CAM) Software, that: - Surveys the stock availability and specifies the raw material to be purchased - Specifies the proper source and controls through bare-codes (complete traceability) - Calculates the needed weights and controls the gravimetric process on the analytical bal- ances - Evaluates the final data received by the in- strumental analyses and calculates the certi- fied values and uncertainties - Creates the labels and certificates - Controls the products intended to be export- ed and prints all accompanying documents. -

Practical Approach to Green Chemistry
International Journal of Pharmacy and Pharmaceutical Sciences ISSN- 0975-1491 Vol 9, Issue 4, 2017 Review Article PRACTICAL APPROACH TO GREEN CHEMISTRY MEENA BHANDARI*, SEEMA RAJ Department of Chemistry, School of Basic and Applied Sciences, K. R. Mangalam University, Sohna Road, Gurgaon, Haryana, India Email: [email protected] Received: 13 Oct 2016 Revised and Accepted: 14 Feb 2017 ABSTRACT The basic principles of green chemistry addresses various issues related to synthesis of chemical compounds: planning organic synthesis to maximise yield, prevention/minimization of waste, atom economy, the use of less lethal chemicals, use of safer solvents, renewable starting materials, energy efficiency and use of green catalysts. The objective of this study is to elaborate the practical approach of green methods. In this paper, we elucidate some important common syntheses having green procedures which can be used in the fields of pharmaceutical chemistry and other fields as well. Green chemistry principles follow up to reduce pollution and environmental degradation by utilizing eco-friendly, non- hazardous, reproducible and efficient solvents and catalysts in the synthesis of drug molecules, drug intermediates and in researches involving synthetic chemistry. The paper also approaches green methods in which microwave radiation can be used as an energy efficient tool. Experimental procedures are gathered from educational journals and laboratory manuals and are viewed in the light of efficacy of green chemistry principles. Keywords: Green chemistry, Organic syntheses, Solvents, Laboratory, Reduce pollution © 2017 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4. -

Product Catalog
For a regularly updated list of pricing and availalble sizes, visit us at www.reagents.com. call: 800-732-8484 fax: 888-843-4384 email: [email protected] 2021 Cover.indd 1 4/26/21 7:55 PM Reagents is your One-Stop Shop Reagents has been a leading manufacturer and distributor of specialty chemicals, reagents, and analytical testing solutions for over 50 years. As Reagents continues to grow, we are constantly adding new brands to our selection. We’ve added a wide selection of lab supply and glassware brands with products that are tested to ASMT guidelines. These Reagents Specialty Reagents Provides: diverse additions allow us to provide a broad portfolio of world class brands manufactured utilizing the highest quality raw Chemicals and Solutions • ISO 17025 Accreditation / ISO 9001 Certified Manufacturing Facility materials meeting or exceeding specifications established by the American Chemical Society. In the laboratory or on the has been a leading • Meets or exceeds stringent manufacturing guidelines production floor, our diverse product portfolio allows consolidation of all of your scientific product needs. manufacturer and • Over 50,000 products available distributor of specialty • Full portfolio of world class brands chemicals, reagents and • High order fill rate within 48 hours on key items analytical testing solutions • Technical and applications support • Degreed and experienced chemical staff since 1969. • Competitive offering and pricing We utilize the highest • Tenured sales specialists with 20+ years of consultative experience quality raw materials to • GHS label compliance meet or exceed the • Many solutions certified traceable to NIST standard reference material specifications established by the American Chemical Custom Solutions: Society.