Development of Chromatography and Mass Spectrometry Methods for Explosives
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ammonium Formate As Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-Ααα-Methylbenzylamine
RSC Advances Ammonium Formate as Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-ααα-Methylbenzylamine Journal: RSC Advances Manuscript ID: RA-ART-01-2014-000462.R1 Article Type: Paper Date Submitted by the Author: 21-Feb-2014 Complete List of Authors: Miranda, Leandro S. M.; Federal University of Rio de Janeiro, Biocatalysis and Organic Synthesis Lab, Chemistry Institute de Souza, Rodrigo Octavio; Federal University of Rio de Janeiro, de Miranda, Amanda; Federal University of Rio de Janeiro, Page 1 of 21 RSC Advances Graphical Abstract RSC Advances Page 2 of 21 Ammonium Formate as Green Hydrogen Source for Clean Semi-Continuous Enzymatic Dynamic Kinetic Resolution of (+/-)-α- Methylbenzylamine Amanda S. de Miranda, [a] Rodrigo O. M. A. de Souza, [ a] Leandro S. M. Miranda [a]* Keywords: Dynamic kinetic resolution • racemic amines • continuous flow . ammonium formate. Abstract: Abstract: The chemoenzymatic dynamic kinetic resolution of (+/-)-α- Methylbenzylamine under continuous flow conditions in the presence of Pd/BaSO 4 as racemization catalyst and ammonium formate as reductant is described. Under the conditions developed good conversions and excellent enantiomeric excess are reported Page 3 of 21 RSC Advances Introduction Recently, continuous processing and biocatalysis have been elected as key green engineering research areas for sustainable manufacturing 1a and it is clear that joint efforts between these areas can lead to great improvements on continuous manufacturing in agreement with green chemistry principles 1b,c . Optically pure amines are ubiquitously present in nature and active pharmaceutical ingredients (APIs). However, their synthesis still represents an ongoing synthetic challenge that can be inferred by the great amount of work and methodologies dealing with this issue in the literature. -

Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives
Article Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives Tomislav Portada, Davor Margetić and Vjekoslav Štrukil * Division of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička cesta 54, 10000 Zagreb, Croatia; [email protected] (T.P.); [email protected] (D.M.) * Correspondence: [email protected]; Tel.: +385‐1‐468‐0197 Received: 15 November 2018; Accepted: 29 November 2018; Published: date Abstract: Mechanochemical ball milling catalytic transfer hydrogenation (CTH) of aromatic nitro compounds using readily available and cheap ammonium formate as the hydrogen source is demonstrated as a simple, facile and clean approach for the synthesis of substituted anilines and selected pharmaceutically relevant compounds. The scope of mechanochemical CTH is broad, as the reduction conditions tolerate various functionalities, for example nitro, amino, hydroxy, carbonyl, amide, urea, amino acid and heterocyclic. The presented methodology was also successfully integrated with other types of chemical reactions previously carried out mechanochemically, such as amide bond formation by coupling amines with acyl chlorides or anhydrides and click‐type coupling reactions between amines and iso(thio)cyanates. In this way, we showed that active pharmaceutical ingredients Procainamide and Paracetamol could be synthesized from the respective nitro‐precursors on milligram and gram scale in excellent isolated yields. Keywords: mechanochemistry; catalytic transfer hydrogenation; aromatic nitro derivatives; ammonium formate; aging; ball milling; synthesis 1. Introduction Catalytic hydrogenation is one of the most significant functional group transformation reactions in organic synthesis and numerous procedures and reagents have been developed for that purpose [1,2]. As such, the hydrogenation reaction plays one of the key roles in many industrially important processes, for example hydrogenation of carbon monoxide to methanol or in food industry for the conversion of unsaturated vegetable oils into saturated triglycerides [3]. -

Deleterious Effects of Formic Acid Without Salt Additives on the HILIC Analysis of Basic Compounds
HPLC TN-1040 Deleterious Effects of Formic Acid without Salt Additives on the HILIC Analysis of Basic Compounds A. Carl Sanchez and Monika Kansal Phenomenex, Inc., Torrance, CA, USA Abstract Formic acid is an often-used mobile phase additive for of weak acids increase. The pKa shifts can be quite significant adjusting pH in reversed phase liquid chromatography (RPLC), in the high organic environment used for HILIC. For example, especially when using mass spectrometric (MS) detection. This weak bases with aqueous pKa less than ~4 typically will not be practice has been carried over to hydrophilic interaction liquid protonated in HILIC mobile phases when 0.1 v/v % formic acid chromatography (HILIC) separations. However, the mechanisms is used. The pKa of the base is decreased in HILIC mobile phase of action and the relative importance of buffer cation and while the pKa of the formic acid is increased. The increased pKa anion are much different in HILIC than RPLC. For this reason of formic acid leads to an increase in mobile phase pH. The buffer selection in HILIC mode requires consideration of buffer, combination of these opposing changes in pKa results in 0.1 v/v analyte and chromatographic sorbent chemical properties to % formic acid being too weak to protonate bases with pKa < ~4. make an appropriate choice. Proper choice of buffer can make Therefore, formic acid can provide acceptable chromatographic the difference between success and failure with HILIC. In this performance for weak bases with aqueous pKa < ~4. However, paper, the behavior of formic acid with and without the addition basic compounds with aqueous pKa greater than ~4 can be of various salts on the HILIC separation of basic analytes is protonated under HILIC conditions with formic acid. -

Nitrate Reductase-Dependent Nitric Oxide Synthesis in the Defense Response of Arabidopsis Thaliana Against Pseudomonas Syringae
Tropical Plant Pathology, vol. 35, 2, 104-107 (2010) Copyright by the Brazilian Phytopathological Society. Printed in Brazil www.sbfito.com.br SHORT COMMUNICATION / COMUNICAÇÃO Nitrate reductase-dependent nitric oxide synthesis in the defense response of Arabidopsis thaliana against Pseudomonas syringae Halley C. Oliveira, Elzira E. Saviani, Jusceley F. P. Oliveira & Ione Salgado Departamento de Biologia Vegetal, Instituto de Biologia, Universidade Estadual de Campinas - UNICAMP, 13083-970, Campinas, SP, Brazil Author for correspondence: Ione Salgado, e-mail: [email protected] ABSTRACT Nitrate reductase (NR) was recently shown to play an important role during phytopathogenic interactions by providing substrates for the synthesis of nitric oxide (NO), a key signal for plant defense responses. In order to give additional support to this hypothesis, we compared NO-mediated defense responses of wild-type and NR double-deficient (nia1 nia2) Arabidopsis thaliana plants inoculated with the IBSBF-1115 (ibs) strain of Pseudomonas syringae pv. maculicola (Psm) and with genetically characterized avirulent (avr) or virulent (vir) strains of Psm. Inoculation of wild-type leaves with avr or ibs, but not vir, stimulated NO emission, as measured by the indicator 4,5-diaminofluorescein. NO emission induced by avr was higher than that induced by ibs. Wild-type plants displayed the hypersensitive response (HR) when infiltrated with the strains avr or ibs, although a stronger HR was induced by avr. The vir strain did not induce HR in wild-type plants, and leaves developed severe infection symptoms. nia1 nia2 plants did not show significantly increased NO emission nor did they develop HR to any of the analyzed strains of Psm, but displayed clorotic lesions and higher bacterial growth in their leaves. -

The Availability of Organic Nitrates from Intravenous Administration Systems
THE AVAILABILITY OF ORGANIC NITRATES FROM INTRAVENOUS ADMINISTRATION SYSTEMS by Paul Adrian Cossum B.Sc., M.P.S. submitted in partial fulfilment of the requirements for the degree of Master of Pharmacy UNIVERSITY OF TASMANIA - HOBART JUNE 1981 SUMMARY Nitroglycerin aind isosorbide dinitrate are two drugs which are infused intravenously during the treatment of ischaemic heart disease. The availability of these two drugs in solutions infused from plastic infusion bags or glass infusion bottles through plastic giving sets has been investigated. During simulated infusions the concentration of nitroglycerin and isosorbide dinitrate appearing in the effluent of the • giving set tubing was found to be much less than the concentration of the drug solution initially contained in the plastic infusion bag or glass infusion bottle. It was found that each component of the plastic infusion equipment sorbed the drugs to a significant extent and that the rate of disappearance of drugs from solutions stored in each component was in the rank order: giving set tubing > giving set burette > plastic infusion bag. There was no significant loss of either drug from solutions stored in glass bottles. The influence of formulation factors and storage conditions on the sorption of nitroglycerin, isosorbide dinitrate and another organic nitrate compound., ethylene glycol dinitrate, by plastic infusion equipment was studied. The extent of loss during simulated infusions was also found to be dependent on flow rate of drug solution through the giving set. The sorption of nitroglycerin and isosorbide dinitrate has clinical and pharmacokinetic significance. Losses of nitroglycerin and isosorbide dinitrate associated with their infusion through plastic Lnfusion equipment were minimised by infusing drug solutions from a glass syringe through high density polyethylene tubing. -

Nitric Oxide Signaling in Plants
plants Editorial Nitric Oxide Signaling in Plants John T. Hancock Department of Applied Sciences, University of the West of England, Bristol BS16 1QY, UK; [email protected]; Tel.: +44-(0)117-328-2475 Received: 3 November 2020; Accepted: 10 November 2020; Published: 12 November 2020 Abstract: Nitric oxide (NO) is an integral part of cell signaling mechanisms in animals and plants. In plants, its enzymatic generation is still controversial. Evidence points to nitrate reductase being important, but the presence of a nitric oxide synthase-like enzyme is still contested. Regardless, NO has been shown to mediate many developmental stages in plants, and to be involved in a range of physiological responses, from stress management to stomatal aperture closure. Downstream from its generation are alterations of the actions of many cell signaling components, with post-translational modifications of proteins often being key. Here, a collection of papers embraces the differing aspects of NO metabolism in plants. Keywords: nitrate reductase; nitration; nitric oxide; reactive oxygen species; stress responses; S-nitrosation; S-nitrosylation; SNO-reductase; thiol modification 1. Introduction Nitric oxide (NO) is now well acknowledged as an instrumental signaling molecule in both plants and animals [1]. First recognized as important as a signal in the control of vascular tone [2], its role in plants came to prominence in the late 1990s [3–5]. The forty years of research into NO in plants has just been highlighted by a review by Kolbert et al. [6]. In plants, NO has been found to be involved in a wide range of developmental stages and physiological responses. -
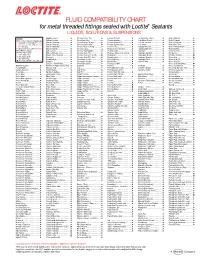
FLUID COMPATIBILITY CHART for Metal Threaded Fittings Sealed with Loctite¨ Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS
FLUID COMPATIBILITY CHART for metal threaded fittings sealed with Loctite® Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS LEGEND: Bagasse Fibers.......................... Chlorobenzene Dry ................... Ferrous Chloride ...................... Ion Exclusion Glycol ................. Nickel Chloride.......................... All Loctite® Anaerobic Sealants are Barium Acetate ........................ Chloroform Dry......................... Ferrous Oxalate......................... Irish Moss Slurry...................... Nickel Cyanide ......................... Compatible Including #242®, 243, Barium Carbonate..................... Chloroformate Methyl............... Ferrous Sulfate10%.................. Iron Ore Taconite ..................... Nickel Fluoborate ..................... 542, 545, 565, 567, 569, 571, 572, Barium Chloride........................ Chlorosulfonic Acid .................. Ferrous Sulfate (Sat)................. Iron Oxide ................................ Nickel Ore Fines ....................... 577, 580, 592 Barium Hydroxide..................... Chrome Acid Cleaning .............. Fertilizer Sol ............................. Isobutyl Alcohol ....................... Nickel Plating Bright ................. † Use Loctite® #270, 271™, 277, 554 Barium Sulfate.......................... Chrome Liquor.......................... Flotation Concentrates.............. Isobutyraldehyde ..................... Nickel Sulfate ........................... Not Recommended Battery Acid .............................. Chrome Plating -

The Noncanonical Pathway for in Vivo Nitric Oxide Generation: the Nitrate-Nitrite-Nitric Oxide Pathway
1521-0081/72/3/692–766$35.00 https://doi.org/10.1124/pr.120.019240 PHARMACOLOGICAL REVIEWS Pharmacol Rev 72:692–766, July 2020 Copyright © 2020 by The Author(s) This is an open access article distributed under the CC BY-NC Attribution 4.0 International license. ASSOCIATE EDITOR: CHRISTOPHER J. GARLAND The Noncanonical Pathway for In Vivo Nitric Oxide Generation: The Nitrate-Nitrite-Nitric Oxide Pathway V. Kapil, R. S. Khambata, D. A. Jones, K. Rathod, C. Primus, G. Massimo, J. M. Fukuto, and A. Ahluwalia William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University London, London, United Kingdom (V.K., R.S.K., D.A.J., K.R., C.P., G.M., A.A.) and Department of Chemistry, Sonoma State University, Rohnert Park, California (J.M.F.) Abstract ...................................................................................694 Significance Statement. ..................................................................694 I. Introduction . ..............................................................................694 A. Chemistry of ·NO and Its Metabolism to Nitrite and Nitrate . .........................695 II. Inorganic Nitrite and Nitrate . ............................................................697 A. Historical Uses of Inorganic Nitrite.....................................................697 B. Historical Uses of Inorganic Nitrate ....................................................698 III. Sources and Pharmacokinetics of Nitrate . ................................................698 -

Dietary Nitrates, Nitrites, and Food Safety: Risks Versus Benefits
Acta Scientific NUTRITIONAL HEALTH (ISSN:2582-1423) Volume 5 Issue 6 June 2021 Review Article Dietary Nitrates, Nitrites, and Food Safety: Risks Versus Benefits Keith R Martin* Received: April 08, 2021 Center for Nutraceutical and Dietary Supplement Research, College of Health Sci- Published: May 26, 2021 ences, University of Memphis, Memphis, USA © All rights are reserved by Keith R Martin. *Corresponding Author: Keith R Martin, Center for Nutraceutical and Dietary Supplement Research, College of Health Sciences, University of Memphis, Memphis, USA. Abstract Nitrates, as a result, are intermediates in the movement of atmospheric nitrogen into the food chain with rich dietary sources includ- Atmospheric nitrogen, via the environmental nitrogen cycle, is captured, or fixed, by symbiotic bacteria interacting with plants. ing red spinach, beetroot, etc. Nitrate-rich fertilizers may further increase nitrogen content of plants. Other sources include potable water, dietary supplements and food additives. Although prevalent in the diet, nitrates have been viewed negatively because they chemically form carcinogenic nitrosamines in acidic environments, e.g. stomach, purportedly leading to gastric cancer as well as neoplasia of the intestine, brain, pancreas, and contributing to Non-Hodgkin’s lymphoma. Other reports indicate associations with hyperthyroidism and diabetes mellitus. A second major concern with dietary nitrate consumption is the development of methemo- - globinemia particularly in infants caused by increases in methemoglobin where -

Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017
EPA/600/R-17/356 | September 2017 www.epa.gov/homeland-security-research Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 Office of Research and Development Homeland Security Research Program This page left intentionally blank EPA/600/R-17/356 | September 2017 Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 UNITED STATES ENVIRONMENTAL PROTECTION AGENCY Cincinnati, OH 45268 Office of Research and Development Homeland Security Research Program Disclaimer Disclaimer The U.S. Environmental Protection Agency (EPA) through its Office of Research and Development funded and managed the research described here under Contract EP-C-15-012 to CSRA Inc. This document is undergoing review and has not been approved for publication. The contents reflect the views of the contributors and technical work groups and do not necessarily reflect the views of the Agency. Mention of trade names or commercial products in this document or in the methods referenced in this document does not constitute endorsement or recommendation for use. Questions concerning this document or its application should be addressed to: Romy Campisano National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King Drive Cincinnati, OH 45268 (513) 569-7016 [email protected] Kathy Hall National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

ATSDR Case Studies in Environmental Medicine Nitrate/Nitrite Toxicity
ATSDR Case Studies in Environmental Medicine Nitrate/Nitrite Toxicity Agency for Toxic Substances and Disease Registry Case Studies in Environmental Medicine (CSEM) Nitrate/Nitrite Toxicity Course: WB2342 CE Original Date: December 5, 2013 CE Renewal Date: December 5, 2015 CE Expiration Date: December 5, 2017 Key • Nitrate toxicity is a preventable cause of Concepts methemoglobinemia. • Infants younger than 4 months of age are at particular risk of nitrate toxicity from contaminated well water. • The widespread use of nitrate fertilizers increases the risk of well-water contamination in rural areas. About This and This educational case study document is one in a series of Other Case self-instructional modules designed to increase the Studies in primary care provider’s knowledge of hazardous Environmental substances in the environment and to promote the Medicine adoption of medical practices that aid in the evaluation and care of potentially exposed patients. The complete series of Case Studies in Environmental Medicine is located on the ATSDR Web site at URL: http://www.atsdr.cdc.gov/csem/csem.html In addition, the downloadable PDF version of this educational series and other environmental medicine materials provides content in an electronic, printable format. Acknowledgements We gratefully acknowledge the work of the medical writers, editors, and reviewers in producing this educational resource. Contributors to this version of the Case Study in Environmental Medicine are listed below. Please Note: Each content expert for this case study has indicated that there is no conflict of interest that would bias the case study content. CDC/ATSDR Author(s): Kim Gehle MD, MPH CDC/ATSDR Planners: Charlton Coles, Ph.D.; Kimberly Gehle, MD; Sharon L.