Gephyrin: a Scaffold That Builds a Phase at the Inhibitory Postsynapses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzymatic Encoding Methods for Efficient Synthesis Of
(19) TZZ__T (11) EP 1 957 644 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12Q 1/68 (2006.01) 01.12.2010 Bulletin 2010/48 C40B 40/06 (2006.01) C40B 50/06 (2006.01) (21) Application number: 06818144.5 (86) International application number: PCT/DK2006/000685 (22) Date of filing: 01.12.2006 (87) International publication number: WO 2007/062664 (07.06.2007 Gazette 2007/23) (54) ENZYMATIC ENCODING METHODS FOR EFFICIENT SYNTHESIS OF LARGE LIBRARIES ENZYMVERMITTELNDE KODIERUNGSMETHODEN FÜR EINE EFFIZIENTE SYNTHESE VON GROSSEN BIBLIOTHEKEN PROCEDES DE CODAGE ENZYMATIQUE DESTINES A LA SYNTHESE EFFICACE DE BIBLIOTHEQUES IMPORTANTES (84) Designated Contracting States: • GOLDBECH, Anne AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DK-2200 Copenhagen N (DK) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • DE LEON, Daen SK TR DK-2300 Copenhagen S (DK) Designated Extension States: • KALDOR, Ditte Kievsmose AL BA HR MK RS DK-2880 Bagsvaerd (DK) • SLØK, Frank Abilgaard (30) Priority: 01.12.2005 DK 200501704 DK-3450 Allerød (DK) 02.12.2005 US 741490 P • HUSEMOEN, Birgitte Nystrup DK-2500 Valby (DK) (43) Date of publication of application: • DOLBERG, Johannes 20.08.2008 Bulletin 2008/34 DK-1674 Copenhagen V (DK) • JENSEN, Kim Birkebæk (73) Proprietor: Nuevolution A/S DK-2610 Rødovre (DK) 2100 Copenhagen 0 (DK) • PETERSEN, Lene DK-2100 Copenhagen Ø (DK) (72) Inventors: • NØRREGAARD-MADSEN, Mads • FRANCH, Thomas DK-3460 Birkerød (DK) DK-3070 Snekkersten (DK) • GODSKESEN, -

And HPV18-Infected Early Stage Cervical Cancers and Normal
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Virology 331 (2005) 269–291 www.elsevier.com/locate/yviro Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: identification of novel candidate molecular markers for cervical cancer diagnosis and therapy Alessandro D. Santina,*, Fenghuang Zhanb, Eliana Bignottia, Eric R. Siegelc, Stefania Cane´ a, Stefania Bellonea, Michela Palmieria, Simone Anfossia, Maria Thomasd, Alexander Burnetta, Helen H. Kaye, Juan J. Romana, Timothy J. O’Briena, Erming Tianb, Martin J. Cannonf, John Shaughnessy Jr.b, Sergio Pecorellig aDivision of Gynecologic Oncology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA bMyeloma Institute for Research and Therapy, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA cDepartment of Biostatistics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA dDepartment of Pathology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA eDepartment of Obstetrics and Gynecology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA fDepartment of Microbiology and Immunology, University of Arkansas, Little Rock, AR 72205, USA gDivision of Gynecologic Oncology, University of Brescia, Brescia, Italy Received 2 July 2004; returned to author for revision 18 August 2004; accepted 9 September 2004 Available online 21 November 2004 Abstract With the goal of identifying genes with a differential pattern of expression between invasive cervical carcinomas (CVX) and normal cervical keratinocytes (NCK), we used oligonucleotide microarrays to interrogate the expression of 14,500 known genes in 11 primary HPV16 and HPV18-infected stage IB–IIA cervical cancers and four primary normal cervical keratinocyte cultures. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
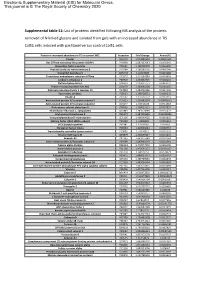
Supplemental Table S1
Electronic Supplementary Material (ESI) for Molecular Omics. This journal is © The Royal Society of Chemistry 2020 Supplemental table S1: List of proteins identified following MS analysis of the proteins removed of N-linked glycans and isolated from gels with an increased abundance in TIS Cal51 cells induced with paclitaxel versus control Cal51 cells. Protein in increased abundance in TIS vs control WCL Accession Fold Change Anova (P) Plectin Q15149 1.073855593 0.00691631 Ras GTPase-activating-like protein IQGAP1 P46940 1.087337643 0.0176342 Elongation factor1-gamma P26641 1.138709703 0.0116496 Peptidyl-prolyl cis-transisomerase B P23284 1.188383105 0.0436246 Dipeptidyl peptidase 3 Q9NY33 1.20163605 0.0215448 Transitional endoplasmic reticulum ATPase P55072 1.214194884 0.0449691 Carbonic anhydrase 2 P00918 1.232852325 0.0158141 Clathrin heavy chain 1 Q00610 1.239621773 0.0463237 Protein transport protein Sec 31A O94979 1.263565104 0.0284155 Aldo-ketoreductase family 1 member C1 Q04828 1.282092186 0.0324406 Spermidine synthase P19623 1.298728621 0.0196232 Plastin-3 P13797 1.310756772 0.0161319 Actin-related protein 2/3 complex subunit 5 O15511 1.333483524 0.00476923 Actin-related protein 2/3 complex subunit 2 O15144 1.35416168 0.0411018 Proteasome subunit alpha type-5 P28066 1.358015551 0.0337657 Thioredoxin reductase 1, cytoplasmic Q16881 1.383670089 0.0235472 Acyl-protein thioesterase 2 O95372 1.387415589 0.00233899 Isoaspartylpeptidase/L-asparaginase Q7L266 1.408149002 0.0319602 Splicing factor U2AF 65kDa subunit P26368 1.41489991 0.0256619 -

Essential Trace Elements in Human Health: a Physician's View
Margarita G. Skalnaya, Anatoly V. Skalny ESSENTIAL TRACE ELEMENTS IN HUMAN HEALTH: A PHYSICIAN'S VIEW Reviewers: Philippe Collery, M.D., Ph.D. Ivan V. Radysh, M.D., Ph.D., D.Sc. Tomsk Publishing House of Tomsk State University 2018 2 Essential trace elements in human health UDK 612:577.1 LBC 52.57 S66 Skalnaya Margarita G., Skalny Anatoly V. S66 Essential trace elements in human health: a physician's view. – Tomsk : Publishing House of Tomsk State University, 2018. – 224 p. ISBN 978-5-94621-683-8 Disturbances in trace element homeostasis may result in the development of pathologic states and diseases. The most characteristic patterns of a modern human being are deficiency of essential and excess of toxic trace elements. Such a deficiency frequently occurs due to insufficient trace element content in diets or increased requirements of an organism. All these changes of trace element homeostasis form an individual trace element portrait of a person. Consequently, impaired balance of every trace element should be analyzed in the view of other patterns of trace element portrait. Only personalized approach to diagnosis can meet these requirements and result in successful treatment. Effective management and timely diagnosis of trace element deficiency and toxicity may occur only in the case of adequate assessment of trace element status of every individual based on recent data on trace element metabolism. Therefore, the most recent basic data on participation of essential trace elements in physiological processes, metabolism, routes and volumes of entering to the body, relation to various diseases, medical applications with a special focus on iron (Fe), copper (Cu), manganese (Mn), zinc (Zn), selenium (Se), iodine (I), cobalt (Co), chromium, and molybdenum (Mo) are reviewed. -

In the MOCS2 Gene
Novel variant (c.472_477del) in the MOCS2 gene Aleksandra Jezela-Stanek1, Witold Blaz2, Artur Gora3, Malgorzata Bochenska2, Katarzyna Kusmierska4, and Jolanta Sykut-Cegielska4 1National Tuberculosis and Lung Diseases Institute 2Saint Jadwiga the Queen Clinical Provincial Hospital No2 3Tunneling Group, Biotechnology Centre, Silesian University of Technology 4Institute of Mother and Child May 26, 2020 Abstract Molybdenum cofactor deficiency type B (MOCODB, #252160) is a rare autosomal recessive metabolic disorder characterized by intractable seizures of neonatal-onset, muscular spasticity, accompanying with hypouricemia, elevated urinary sulfite levels and craniofacial dysmorphism. Thirty-five patients were reported to date. Our paper aimed to delineate the disease genotype by presenting another patient, in whom novel, inframe variant within the MOCS2 gene was identified. Its clinical significance was supported by the medical history and analysis of the possible mutation consequences on a molecular level with the use of the available crystal structure of the human molybdopterin synthase complex. Moreover, potential pathomechanism resulting from molecular defect was presented, giving original insight into current knowledge on this rare disease, including treatment options. Introduction Molybdenum cofactor deficiency type B (MOCODB, #252160) is an autosomal recessive metabolic dis- order characterized by intractable seizures of neonatal-onset, muscular spasticity, accompanying with hy- pouricemia, elevated urinary sulfite levels and craniofacial dysmorphism. It came to medical attention first in 1980 (Johnson, 1980). Affected children show severe neurologic complications, which may lead to early death, rarely (only three cases described to date) presented with a milder form with global developmen- tal delay without seizures (Arican, 2019). The disorder results from decreased activity of sulfite oxidase (SUOX; EC 1.8.3.1) and xanthine dehydrogenase (XDH; EC 1.17.1.4 and 1.17.3.2), which are molybdenum cofactor-dependent for their activity. -

Structural and Biochemical Characterization of Gephyrin and Various Gephyrin-Ligand Complexes
Structural and biochemical characterization of gephyrin and various gephyrin-ligand complexes Strukturelle und biochemische Charakterisierung von Gephyrin und verschiedenen Gephyrin-Liganden- Komplexen Doctoral thesis for a doctoral degree at the Graduate School of Life Sciences, Julius-Maximilians-Universität Würzburg, Section Biomedicine Submitted by Bodo Sander from Frankfurt am Main Würzburg, 30th of May 2014 Dedicated to my dear Gudrun Submitted on: ………………………………………………………..…….. Office stamp Members of the Promotionskomitee: Chairperson: Thomas Dandekar Primary Supervisor: Hermann Schindelin Supervisor (Second): Thomas Müller Supervisor (Third): Thomas Raabe Date of Public Defense: ………………………………………………………..…….. Date of Receipt of Certificates: ………………………………………………………..…….. TABLE OF CONTENTS 3 TABLE OF CONTENTS TABLE OF CONTENTS .................................................................................................................... 3 AFFIDAVIT/EIDESSTATTLICHE ERKLÄRUNG .................................................................................. 11 ACKNOWLEDGEMENTS ............................................................................................................... 12 SUMMARY ................................................................................................................................. 14 ZUSAMMENFASSUNG ................................................................................................................. 17 I. MAIN INTRODUCTION .......................................................................................................... -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Clinical and Biochemical Spectrum of Molybdenum Cofactor Deficiency
Research PERINATOLOGY • Vol 20 • No. 4 • Jan–Mar 2020 Article Clinical and Biochemical Spectrum of Molybdenum Cofactor Deficiency Due To MOCS2 Mutations Ketki Vinod Kudalkar, Arndt Rolfs, Elham Kashani, Christian Beetz, Manish Parakh, Ravikumar Sowmya, Chinthalapalli Prakash Ravi Kumar, Anil Bansidhar Jalan* Abstract Background: Molybdenum cofactor deficiency (MoCD) is a neurometabolic disorder with presenting symptoms such as severe congenital microcephaly, severe global developmen- tal delay, intractable seizure disorder, and spastic quadriple- gia. Magnetic resonance imaging of the brain of patients with MoCD indicates brain atrophy, delayed myelination, and cystic leukomalacia. Materials and Methods: We evaluated 3 patients with MoCD and their clinical, biochemical, and molecular findings. The results were compared with previously reported cases. One of these patients was prescribed a low-methionine diet, and the clinical and biochemical changes observed in this case are *Correspondence presented in this article. Dr Anil Bansidhar Jalan Results: In all 3 patients with MoCD, uric acid and homocyst- Chief Scientific Research Officer eine levels were low, and sulfocysteine, urinary hypoxan- Division of Biochemical Genetics thine, and xanthine levels were elevated. We also noticed Navi Mumbai Institute of Research in Mental and homozygous mutations in the MOCS2 gene of these patients. Neurological Handicap Methionine-restricted diet in 1 patient with a milder mutation C-116, Om Rachna Society, Sector 17, Vashi showed good clinical response with improvement in head Navi Mumbai 400705, Maharashtra control and reduced frequency of seizures. Biochemical inves- India tigations showed improvement in decreased sulfocysteine E-mail: [email protected] level in the plasma and urine along with disappearance of sulfites in the urine. -

Gephyrin: a Central Gabaergic Synapse Organizer
OPEN Experimental & Molecular Medicine (2015) 47, e158; doi:10.1038/emm.2015.5 & 2015 KSBMB. All rights reserved 2092-6413/15 www.nature.com/emm REVIEW Gephyrin: a central GABAergic synapse organizer Gayoung Choii1 and Jaewon Ko1,2 Gephyrin is a central element that anchors, clusters and stabilizes glycine and γ-aminobutyric acid type A receptors at inhibitory synapses of the mammalian brain. It self-assembles into a hexagonal lattice and interacts with various inhibitory synaptic proteins. Intriguingly, the clustering of gephyrin, which is regulated by multiple posttranslational modifications, is critical for inhibitory synapse formation and function. In this review, we summarize the basic properties of gephyrin and describe recent findings regarding its roles in inhibitory synapse formation, function and plasticity. We will also discuss the implications for the pathophysiology of brain disorders and raise the remaining open questions in this field. Experimental & Molecular Medicine (2015) 47, e158; doi:10.1038/emm.2015.5; published online 17 April 2015 INTRODUCTION Gephyrin is the most extensively studied scaffold responsible Synapses, which are the fundamental information-processing for organizing the inhibitory postsynaptic density, which is units of neural circuits, form the basis for all brain functions by essential for clustering of glycine and γ-aminobutyric acid type controlling the excitation-to-inhibition balance. Synapses form A (GABAA) receptors, inhibitory synaptic transmission and via a coordinated process that orchestrates (sequentially or in long-term potentiation (reviewed in refs 4–6). Since the parallel) a variety of dynamically regulated cellular events. discovery of gephyrin in 1982 by Heinrich Betz’sgroup,7 its Various molecular and cellular mechanisms underlying synapse biochemical and cellular properties have been extensively formation have been elucidated. -

Emerging Common Molecular Pathways for Primary Dystonia
REVIEW Emerging Common Molecular Pathways for Primary Dystonia Mark S. LeDoux, MD, PhD,1 William T. Dauer, MD,2 and Thomas T. Warner, FRCP, PhD3* 1Department of Neurology, University of Tennessee Health Science Center Memphis, Tennessee 38163, USA 2Department of Neurology and Cell & Developmental Biology, University of Michigan Medical School, Ann Arbor, Michigan, USA 3Reta Lila Weston Institute for Neurological Research, UCL Institute of Neurology, London, United Kingdom ABSTRACT: The dystonias are a group of English-language publications available on PubMed hyperkinetic movement disorders whose principal cause relating to the genetics and cellular pathology of dysto- is neuron dysfunction at 1 or more interconnected nia was performed. Numerous potential pathogenetic nodes of the motor system. The study of genes and mechanisms have been identified. We describe those proteins that cause familial dystonia provides critical in- that fall into 3 emerging thematic groups: cell-cycle and formation about the cellular pathways involved in this transcriptional regulation in the nucleus, endoplasmic dysfunction, which disrupts the motor pathways at the reticulum and nuclear envelope function, and control of systems level. In recent years study of the increasing synaptic function. VC 2013 Movement Disorder Society number of DYT genes has implicated a number of cell functions that appear to be involved in the pathogene- Key Words: DYT genes; cell cycle; endoplasmic sis of dystonia. A review of the literature published in reticulum; nuclear envelope; synaptic function The dystonias are a heterogeneous group of hyperki- (CNS) damage and the dystonic movements are part netic movement disorders characterized by sustained of a more complex neurological phenotype.