In the MOCS2 Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzymatic Encoding Methods for Efficient Synthesis Of
(19) TZZ__T (11) EP 1 957 644 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12Q 1/68 (2006.01) 01.12.2010 Bulletin 2010/48 C40B 40/06 (2006.01) C40B 50/06 (2006.01) (21) Application number: 06818144.5 (86) International application number: PCT/DK2006/000685 (22) Date of filing: 01.12.2006 (87) International publication number: WO 2007/062664 (07.06.2007 Gazette 2007/23) (54) ENZYMATIC ENCODING METHODS FOR EFFICIENT SYNTHESIS OF LARGE LIBRARIES ENZYMVERMITTELNDE KODIERUNGSMETHODEN FÜR EINE EFFIZIENTE SYNTHESE VON GROSSEN BIBLIOTHEKEN PROCEDES DE CODAGE ENZYMATIQUE DESTINES A LA SYNTHESE EFFICACE DE BIBLIOTHEQUES IMPORTANTES (84) Designated Contracting States: • GOLDBECH, Anne AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DK-2200 Copenhagen N (DK) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • DE LEON, Daen SK TR DK-2300 Copenhagen S (DK) Designated Extension States: • KALDOR, Ditte Kievsmose AL BA HR MK RS DK-2880 Bagsvaerd (DK) • SLØK, Frank Abilgaard (30) Priority: 01.12.2005 DK 200501704 DK-3450 Allerød (DK) 02.12.2005 US 741490 P • HUSEMOEN, Birgitte Nystrup DK-2500 Valby (DK) (43) Date of publication of application: • DOLBERG, Johannes 20.08.2008 Bulletin 2008/34 DK-1674 Copenhagen V (DK) • JENSEN, Kim Birkebæk (73) Proprietor: Nuevolution A/S DK-2610 Rødovre (DK) 2100 Copenhagen 0 (DK) • PETERSEN, Lene DK-2100 Copenhagen Ø (DK) (72) Inventors: • NØRREGAARD-MADSEN, Mads • FRANCH, Thomas DK-3460 Birkerød (DK) DK-3070 Snekkersten (DK) • GODSKESEN, -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Gephyrin: a Scaffold That Builds a Phase at the Inhibitory Postsynapses
www.nature.com/cr www.cell-research.com RESEARCH HIGHLIGHT OPEN Gephyrin: a scaffold that builds a phase at the inhibitory postsynapses Christian Hoffmann 1 and Dragomir Milovanovic 1 Cell Research (2021) 31:245–246; https://doi.org/10.1038/s41422-020-00440-2 The scaffolding protein gephyrin is a core component of many GPHN-E. Besides GlyR-β, TM3–4 loops of GABAAR α3-subunit also inhibitory synapses. Bai and colleagues now show that phase phase separates with GPHN-E suggesting liquid condensation to separation of gephyrin with the subunits of both glycine and be a general feature of inhibitory synapses (Fig. 1). The phase GABA receptors underlies the formation of postsynaptic separation occurs at the equimolar ratio of receptors and scaffold sheets at the inhibitory synapses. proteins, a range similar to their physiological concentrations.8 To assure precise processing of information, contacts between There are several implications of this study. First, the observa- neurons require the alignment of the release sites, presynaptic tion that different regions of the same proteins (GlyR-β and GPHN- terminals, with the ionotropic transmitter receptors at the E) are important for phase separation or direct binding, suggests postsynaptic plasma membrane. Downstream of these receptors, that these two processes could be regulated separately. The scaffold molecules, kinases and cytoskeleton components all positively charged region of GlyR-β (a.a. 354–383) and negatively orchestrate the signaling cascade induced by the neurotransmitter charged surface at the subdomain II of GPHN-E are essential for fl 1234567890();,: binding and ion in ux. Each neuron in the mammalian central condensate formation, despite hydrophobic interactions being nervous system relies on both the excitatory and inhibitory inputs. -

And HPV18-Infected Early Stage Cervical Cancers and Normal
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Virology 331 (2005) 269–291 www.elsevier.com/locate/yviro Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: identification of novel candidate molecular markers for cervical cancer diagnosis and therapy Alessandro D. Santina,*, Fenghuang Zhanb, Eliana Bignottia, Eric R. Siegelc, Stefania Cane´ a, Stefania Bellonea, Michela Palmieria, Simone Anfossia, Maria Thomasd, Alexander Burnetta, Helen H. Kaye, Juan J. Romana, Timothy J. O’Briena, Erming Tianb, Martin J. Cannonf, John Shaughnessy Jr.b, Sergio Pecorellig aDivision of Gynecologic Oncology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA bMyeloma Institute for Research and Therapy, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA cDepartment of Biostatistics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA dDepartment of Pathology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA eDepartment of Obstetrics and Gynecology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA fDepartment of Microbiology and Immunology, University of Arkansas, Little Rock, AR 72205, USA gDivision of Gynecologic Oncology, University of Brescia, Brescia, Italy Received 2 July 2004; returned to author for revision 18 August 2004; accepted 9 September 2004 Available online 21 November 2004 Abstract With the goal of identifying genes with a differential pattern of expression between invasive cervical carcinomas (CVX) and normal cervical keratinocytes (NCK), we used oligonucleotide microarrays to interrogate the expression of 14,500 known genes in 11 primary HPV16 and HPV18-infected stage IB–IIA cervical cancers and four primary normal cervical keratinocyte cultures. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
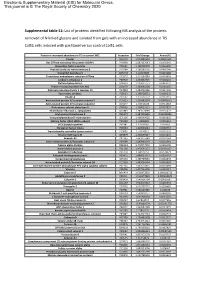
Supplemental Table S1
Electronic Supplementary Material (ESI) for Molecular Omics. This journal is © The Royal Society of Chemistry 2020 Supplemental table S1: List of proteins identified following MS analysis of the proteins removed of N-linked glycans and isolated from gels with an increased abundance in TIS Cal51 cells induced with paclitaxel versus control Cal51 cells. Protein in increased abundance in TIS vs control WCL Accession Fold Change Anova (P) Plectin Q15149 1.073855593 0.00691631 Ras GTPase-activating-like protein IQGAP1 P46940 1.087337643 0.0176342 Elongation factor1-gamma P26641 1.138709703 0.0116496 Peptidyl-prolyl cis-transisomerase B P23284 1.188383105 0.0436246 Dipeptidyl peptidase 3 Q9NY33 1.20163605 0.0215448 Transitional endoplasmic reticulum ATPase P55072 1.214194884 0.0449691 Carbonic anhydrase 2 P00918 1.232852325 0.0158141 Clathrin heavy chain 1 Q00610 1.239621773 0.0463237 Protein transport protein Sec 31A O94979 1.263565104 0.0284155 Aldo-ketoreductase family 1 member C1 Q04828 1.282092186 0.0324406 Spermidine synthase P19623 1.298728621 0.0196232 Plastin-3 P13797 1.310756772 0.0161319 Actin-related protein 2/3 complex subunit 5 O15511 1.333483524 0.00476923 Actin-related protein 2/3 complex subunit 2 O15144 1.35416168 0.0411018 Proteasome subunit alpha type-5 P28066 1.358015551 0.0337657 Thioredoxin reductase 1, cytoplasmic Q16881 1.383670089 0.0235472 Acyl-protein thioesterase 2 O95372 1.387415589 0.00233899 Isoaspartylpeptidase/L-asparaginase Q7L266 1.408149002 0.0319602 Splicing factor U2AF 65kDa subunit P26368 1.41489991 0.0256619 -

Essential Trace Elements in Human Health: a Physician's View
Margarita G. Skalnaya, Anatoly V. Skalny ESSENTIAL TRACE ELEMENTS IN HUMAN HEALTH: A PHYSICIAN'S VIEW Reviewers: Philippe Collery, M.D., Ph.D. Ivan V. Radysh, M.D., Ph.D., D.Sc. Tomsk Publishing House of Tomsk State University 2018 2 Essential trace elements in human health UDK 612:577.1 LBC 52.57 S66 Skalnaya Margarita G., Skalny Anatoly V. S66 Essential trace elements in human health: a physician's view. – Tomsk : Publishing House of Tomsk State University, 2018. – 224 p. ISBN 978-5-94621-683-8 Disturbances in trace element homeostasis may result in the development of pathologic states and diseases. The most characteristic patterns of a modern human being are deficiency of essential and excess of toxic trace elements. Such a deficiency frequently occurs due to insufficient trace element content in diets or increased requirements of an organism. All these changes of trace element homeostasis form an individual trace element portrait of a person. Consequently, impaired balance of every trace element should be analyzed in the view of other patterns of trace element portrait. Only personalized approach to diagnosis can meet these requirements and result in successful treatment. Effective management and timely diagnosis of trace element deficiency and toxicity may occur only in the case of adequate assessment of trace element status of every individual based on recent data on trace element metabolism. Therefore, the most recent basic data on participation of essential trace elements in physiological processes, metabolism, routes and volumes of entering to the body, relation to various diseases, medical applications with a special focus on iron (Fe), copper (Cu), manganese (Mn), zinc (Zn), selenium (Se), iodine (I), cobalt (Co), chromium, and molybdenum (Mo) are reviewed. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Molecular Diagnostic Requisition
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 MOLECULAR DIAGNOSTIC REQUISITION PATIENT INFORMATION SAMPLE INFORMATION NAME: DATE OF COLLECTION: / / LAST NAME FIRST NAME MI MM DD YY HOSPITAL#: ACCESSION#: DATE OF BIRTH: / / GENDER (Please select one): FEMALE MALE MM DD YY SAMPLE TYPE (Please select one): ETHNIC BACKGROUND (Select all that apply): UNKNOWN BLOOD AFRICAN AMERICAN CORD BLOOD ASIAN SKELETAL MUSCLE ASHKENAZIC JEWISH MUSCLE EUROPEAN CAUCASIAN -OR- DNA (Specify Source): HISPANIC NATIVE AMERICAN INDIAN PLACE PATIENT STICKER HERE OTHER JEWISH OTHER (Specify): OTHER (Please specify): REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: INDICATION FOR STUDY SYMPTOMATIC (Summarize below.): *FAMILIAL MUTATION/VARIANT ANALYSIS: COMPLETE ALL FIELDS BELOW AND ATTACH THE PROBAND'S REPORT. GENE NAME: ASYMPTOMATIC/POSITIVE FAMILY HISTORY: (ATTACH FAMILY HISTORY) MUTATION/UNCLASSIFIED VARIANT: RELATIONSHIP TO PROBAND: THIS INDIVIDUAL IS CURRENTLY: SYMPTOMATIC ASYMPTOMATIC *If family mutation is known, complete the FAMILIAL MUTATION/ VARIANT ANALYSIS section. NAME OF PROBAND: ASYMPTOMATIC/POPULATION SCREENING RELATIONSHIP TO PROBAND: OTHER (Specify clinical findings below): BMGL LAB#: A COPY OF ORIGINAL RESULTS ATTACHED IF PROBAND TESTING WAS PERFORMED AT ANOTHER LAB, CALL TO DISCUSS PRIOR TO SENDING SAMPLE. A POSITIVE CONTROL MAY BE REQUIRED IN SOME CASES. REQUIRED: NEW YORK STATE PHYSICIAN SIGNATURE OF CONSENT I certify that the patient specified above and/or their legal guardian has been informed of the benefits, risks, and limitations of the laboratory test(s) requested. -

Blueprint Genetics MOCS2 Single Gene Test
MOCS2 single gene test Test code: S01194 Phenotype information Molybdenum cofactor deficiency Alternative gene names MOCO1 Panels that include the MOCS2 gene Lysosomal Disorders and Mucopolysaccharidosis Panel Comprehensive Metabolism Panel Test Strengths The strengths of this test include: CAP accredited laboratory CLIA-certified personnel performing clinical testing in a CLIA-certified laboratory Powerful sequencing technologies, advanced target enrichment methods and precision bioinformatics pipelines ensure superior analytical performance Careful construction of clinically effective and scientifically justified gene panels Our Nucleus online portal providing transparent and easy access to quality and performance data at the patient level Our publicly available analytic validation demonstrating complete details of test performance ~2,000 non-coding disease causing variants in our clinical grade NGS assay for panels (please see ‘Non-coding disease causing variants covered by this test’) Our rigorous variant classification scheme Our systematic clinical interpretation workflow using proprietary software enabling accurate and traceable processing of NGS data Our comprehensive clinical statements Test Limitations This test does not detect the following: Complex inversions Gene conversions Balanced translocations Mitochondrial DNA variants Repeat expansion disorders unless specifically mentioned Non-coding variants deeper than ±20 base pairs from exon-intron boundary unless otherwise indicated (please see above non-coding variants covered by the test). This test may not reliably detect the following: Low level mosaicism (variant with a minor allele fraction of 14.6% is detected with 90% probability) Stretches of mononucleotide repeats Indels larger than 50bp Single exon deletions or duplications https://blueprintgenetics.com/ Variants within pseudogene regions/duplicated segments The sensitivity of this test may be reduced if DNA is extracted by a laboratory other than Blueprint Genetics. -

Citrullinemia Type I Is Associated with a Novel
Lin et al. BMC Medical Genetics (2019) 20:110 https://doi.org/10.1186/s12881-019-0836-5 CASE REPORT Open Access Citrullinemia type I is associated with a novel splicing variant, c.773 + 4A > C, in ASS1: a case report and literature review Yiming Lin1, Hongzhi Gao2, Bin Lu3, Shuang Zhou2, Tianwen Zheng4, Weihua Lin1, Lin Zhu3, Mengyi Jiang3* and Qingliu Fu1* Abstract Background: Citrullinemia type I (CTLN1) is a rare autosomal recessive disorder of the urea cycle caused by a deficiency in the argininosuccinate synthetase (ASS1) enzyme due to mutations in the ASS1 gene. Only a few Chinese patients with CTLN1 have been reported, and ASS1 gene mutations have been identified sporadically in China. Case presentation: A Chinese family with one member affected with mild CTLN1 was enrolled. Targeted exome sequencing was performed on the proband, and Sanger sequencing was used to validate the detected mutation. We also reviewed the genetic and clinical characteristics of CTLN1 in Chinese patients that have been published to date. Newborn screening showed remarkably increased concentrations of citrulline with elevated ratios of citrulline/arginine and citrulline/phenylalanine, and the patient presented with a speech delay at age three. The urinary organic acid profiles were normal. A novel homozygous splicing variant c.773 + 4A > C in the ASS1 gene was identified in the proband, and it was predicted to affect splicing by in silico analysis. To date, only nine Chinese patients with CTLN1 have been reported, with a total of 15 ASS1 mutations identified and no high frequency or hot spot mutations found; the mutation spectrum of Chinese patients with CTLN1 was heterogeneous. -

Structural and Biochemical Characterization of Gephyrin and Various Gephyrin-Ligand Complexes
Structural and biochemical characterization of gephyrin and various gephyrin-ligand complexes Strukturelle und biochemische Charakterisierung von Gephyrin und verschiedenen Gephyrin-Liganden- Komplexen Doctoral thesis for a doctoral degree at the Graduate School of Life Sciences, Julius-Maximilians-Universität Würzburg, Section Biomedicine Submitted by Bodo Sander from Frankfurt am Main Würzburg, 30th of May 2014 Dedicated to my dear Gudrun Submitted on: ………………………………………………………..…….. Office stamp Members of the Promotionskomitee: Chairperson: Thomas Dandekar Primary Supervisor: Hermann Schindelin Supervisor (Second): Thomas Müller Supervisor (Third): Thomas Raabe Date of Public Defense: ………………………………………………………..…….. Date of Receipt of Certificates: ………………………………………………………..…….. TABLE OF CONTENTS 3 TABLE OF CONTENTS TABLE OF CONTENTS .................................................................................................................... 3 AFFIDAVIT/EIDESSTATTLICHE ERKLÄRUNG .................................................................................. 11 ACKNOWLEDGEMENTS ............................................................................................................... 12 SUMMARY ................................................................................................................................. 14 ZUSAMMENFASSUNG ................................................................................................................. 17 I. MAIN INTRODUCTION ..........................................................................................................