Copy Number Variation Study in Japanese Quail Associated with Stress Related Traits Using Whole Genome Re-Sequencing Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
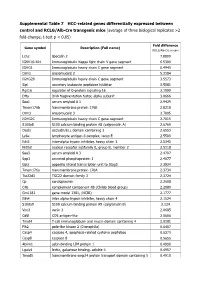
Supplemental Table 7 HCC-Related Genes Differentially Expressed Between Control and RCLG/Alb-Cre Transgenic Mice
Supplemental Table 7 HCC-related genes differentially expressed between control and RCLG/Alb-Cre transgenic mice (average of three biological replicates >2 fold-change, t-test p < 0.05) Fold difference Gene symbol Description (Full name) (RCLG/Alb-Cre vs con) Lcn2 lipocalin 2 7.8899 IGKV16-104 Immunoglobulin Kappa light chain V gene segment 6.5300 IGHG1 Immunoglobulin heavy chain C gene segment 6.4945 Orm2 orosomucoid 2 5.3184 IGHG2B Immunoglobulin heavy chain C gene segment 3.5573 Slpi secretory leukocyte peptidase inhibitor 3.5081 Rgs16 regulator of G-protein signaling 16 3.1999 Dffa DNA fragmentation factor, alpha subunit 3.0666 Saa1 serum amyloid A 1 2.9429 Tmem176b transmembrane protein 176B 2.8218 Orm3 orosomucoid 3 2.7805 IGHG2C Immunoglobulin heavy chain C gene segment 2.7015 S100a8 S100 calcium binding protein A8 (calgranulin A) 2.6769 Ocel1 occludin/ELL domain containing 1 2.6553 Ly6e lymphocyte antigen 6 complex, locus E 2.5509 Itih3 inter-alpha trypsin inhibitor, heavy chain 3 2.5345 Nr0b2 nuclear receptor subfamily 0, group B, member 2 2.5118 Saa3 serum amyloid A 3 2.4707 Spp1 secreted phosphoprotein 1 2.4077 Gats opposite strand transcription unit to Stag3 2.3934 Tmem176a transmembrane protein 176A 2.3734 Tsc22d3 TSC22 domain family 3 2.3724 Cp ceruloplasmin 2.2608 C4b complement component 4B (Childo blood group) 2.2089 Gm1381 gene model 1381, (NCBI) 2.1777 Itih4 inter alpha-trypsin inhibitor, heavy chain 4 2.1524 S100a9 S100 calcium binding protein A9 (calgranulin B) 2.124 Vnn3 vanin 3 2.0685 Cd5l CD5 antigen-like 2.0606 -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Looking for Missing Proteins in the Proteome Of
Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update Yves Vandenbrouck, Lydie Lane, Christine Carapito, Paula Duek, Karine Rondel, Christophe Bruley, Charlotte Macron, Anne Gonzalez de Peredo, Yohann Coute, Karima Chaoui, et al. To cite this version: Yves Vandenbrouck, Lydie Lane, Christine Carapito, Paula Duek, Karine Rondel, et al.. Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update. Journal of Proteome Research, American Chemical Society, 2016, 15 (11), pp.3998-4019. 10.1021/acs.jproteome.6b00400. hal-02191502 HAL Id: hal-02191502 https://hal.archives-ouvertes.fr/hal-02191502 Submitted on 19 Mar 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Journal of Proteome Research 1 2 3 Looking for missing proteins in the proteome of human spermatozoa: an 4 update 5 6 Yves Vandenbrouck1,2,3,#,§, Lydie Lane4,5,#, Christine Carapito6, Paula Duek5, Karine Rondel7, 7 Christophe Bruley1,2,3, Charlotte Macron6, Anne Gonzalez de Peredo8, Yohann Couté1,2,3, 8 Karima Chaoui8, Emmanuelle Com7, Alain Gateau5, AnneMarie Hesse1,2,3, Marlene 9 Marcellin8, Loren Méar7, Emmanuelle MoutonBarbosa8, Thibault Robin9, Odile Burlet- 10 Schiltz8, Sarah Cianferani6, Myriam Ferro1,2,3, Thomas Fréour10,11, Cecilia Lindskog12,Jérôme 11 1,2,3 7,§ 12 Garin , Charles Pineau . -

The Roles of RNA Polymerase I and III Subunits Polr1a, Polr1c, and Polr1d in Craniofacial Development BY
The roles of RNA Polymerase I and III subunits Polr1a, Polr1c, and Polr1d in craniofacial development BY © 2016 Kristin Emily Noack Watt Submitted to the graduate degree program in Anatomy and Cell Biology and to the Graduate Faculty of The University of Kansas Medical Center in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Brian Andrews Jennifer Gerton Tatjana Piotrowski Russell Swerdlow Date Defended: January 26, 2016 The Dissertation Committee for Kristin Watt certifies that this is the approved version of the following dissertation: The roles of RNA Polymerase I and III subunits Polr1a, Polr1c, and Polr1d in craniofacial development Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Date approved: February 2, 2016 ii Abstract Craniofacial anomalies account for approximately one-third of all birth defects. Two examples of syndromes associated with craniofacial malformations are Treacher Collins syndrome and Acrofacial Dysostosis, Cincinnati type which have phenotypic overlap including deformities of the eyes, ears, and facial bones. Mutations in TCOF1, POLR1C or POLR1D may cause Treacher Collins syndrome while mutations in POLR1A may cause Acrofacial Dysostosis, Cincinnati type. TCOF1 encodes the nucleolar phosphoprotein Treacle, which functions in rRNA transcription and modification. Previous studies demonstrated that Tcof1 mutations in mice result in reduced ribosome biogenesis and increased neuroepithelial apoptosis. This diminishes the neural crest cell (NCC) progenitor population which contribute to the development of the cranial skeleton. In contrast, apart from being subunits of RNA Polymerases (RNAP) I and/or III, nothing is known about the function of POLR1A, POLR1C, and POLR1D during embryonic and craniofacial development. -
![Downloaded the CDS Fasta File and Gff File from the Ensembl Ftp Server (Release-98) [39]](https://docslib.b-cdn.net/cover/6641/downloaded-the-cds-fasta-file-and-gff-file-from-the-ensembl-ftp-server-release-98-39-1196641.webp)
Downloaded the CDS Fasta File and Gff File from the Ensembl Ftp Server (Release-98) [39]
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.25.266486; this version posted April 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Frequent lineage-specific substitution rate changes support an episodic model for protein evolution Neel Prabh1,2 and Diethard Tautz1 1. Department of Evolutionary Genetics, Max Planck Institute for Evolutionary Biology, August-Thienemann-Str. 2, 24306 Plön, Germany 2. Corresponding author E-mail: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.08.25.266486; this version posted April 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 2 Abstract Since the inception of the molecular clock model for sequence evolution, the investigation of protein divergence has revolved around the question of a more or less constant rate of overall sequence information change. Although anomalies in clock-like divergence are described for some proteins, nowadays, the assumption of a constant decay rate for a given protein family is taken as the null model for protein evolution. Still, so far, a systematic test of this null model has not been done at a genome-wide scale despite the databases’ enormous growth. We focus here on divergence rate comparisons between closely related lineages, since this allows clear orthology assignments by synteny and unequivocal alignments, which are crucial for the determination of substitution rate changes. Thus, we generated a high-confidence dataset of syntenic orthologs from four ape species, including humans. -

MOCHI Enables Discovery of Heterogeneous Interactome Modules in 3D Nucleome
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press MOCHI enables discovery of heterogeneous interactome modules in 3D nucleome Dechao Tian1,# , Ruochi Zhang1,# , Yang Zhang1, Xiaopeng Zhu1, and Jian Ma1,* 1Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA #These two authors contributed equally *Correspondence: [email protected] Contact To whom correspondence should be addressed: Jian Ma School of Computer Science Carnegie Mellon University 7705 Gates-Hillman Complex 5000 Forbes Avenue Pittsburgh, PA 15213 Phone: +1 (412) 268-2776 Email: [email protected] 1 Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The composition of the cell nucleus is highly heterogeneous, with different constituents forming complex interactomes. However, the global patterns of these interwoven heterogeneous interactomes remain poorly understood. Here we focus on two different interactomes, chromatin interaction network and gene regulatory network, as a proof-of-principle, to identify heterogeneous interactome modules (HIMs), each of which represents a cluster of gene loci that are in spatial contact more frequently than expected and that are regulated by the same group of transcription factors. HIM integrates transcription factor binding and 3D genome structure to reflect “transcriptional niche” in the nucleus. We develop a new algorithm MOCHI to facilitate the discovery of HIMs based on network motif clustering in heterogeneous interactomes. By applying MOCHI to five different cell types, we found that HIMs have strong spatial preference within the nucleus and exhibit distinct functional properties. Through integrative analysis, this work demonstrates the utility of MOCHI to identify HIMs, which may provide new perspectives on the interplay between transcriptional regulation and 3D genome organization. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Qt38n028mr Nosplash A3e1d84
! ""! ACKNOWLEDGEMENTS I dedicate this thesis to my parents who inspired me to become a scientist through invigorating scientific discussions at the dinner table even when I was too young to understand what the hippocampus was. They also prepared me for the ups and downs of science and supported me through all of these experiences. I would like to thank my advisor Dr. Elizabeth Blackburn and my thesis committee members Dr. Eric Verdin, and Dr. Emmanuelle Passegue. Liz created a nurturing and supportive environment for me to explore my own ideas, while at the same time teaching me how to love science, test my questions, and of course provide endless ways to think about telomeres and telomerase. Eric and Emmanuelle both gave specific critical advice about the proper experiments for T cells and both volunteered their lab members for further critical advice. I always felt inspired with a sense of direction after thesis committee meetings. The Blackburn lab is full of smart and dedicated scientists whom I am thankful for their support. Specifically Dr. Shang Li and Dr. Brad Stohr for their stimulating scientific debates and “arguments.” Dr. Jue Lin, Dana Smith, Kyle Lapham, Dr. Tet Matsuguchi, and Kyle Jay for their friendships and discussions about what my data could possibly mean. Dr. Eva Samal for teaching me molecular biology techniques and putting up with my late night lab exercises. Beth Cimini for her expertise with microscopy, FACs, singing, and most of all for being a caring and supportive friend. Finally, I would like to thank Dr. Imke Listerman, my scientific partner for most of the breast cancer experiments. -

The Genetic Basis of Malformation of Cortical Development Syndromes: Primary Focus on Aicardi Syndrome
The Genetic Basis of Malformation of Cortical Development Syndromes: Primary Focus on Aicardi Syndrome Thuong Thi Ha B. Sc, M. Bio Neurogenetics Research Group The University of Adelaide Thesis submitted for the degree of Doctor of Philosophy In Discipline of Genetics and Evolution School of Biological Sciences Faculty of Science The University of Adelaide June 2018 Table of Contents Abstract 6 Thesis Declaration 8 Acknowledgements 9 Publications 11 Abbreviations 12 CHAPTER I: Introduction 15 1.1 Overview of Malformations of Cortical Development (MCD) 15 1.2 Introduction into Aicardi Syndrome 16 1.3 Clinical Features of Aicardi Syndrome 20 1.3.1 Epidemiology 20 1.3.2 Clinical Diagnosis 21 1.3.3 Differential Diagnosis 23 1.3.4 Development & Prognosis 24 1.4 Treatment 28 1.5 Pathogenesis of Aicardi Syndrome 29 1.5.1 Prenatal or Intrauterine Disturbances 29 1.5.2 Genetic Predisposition 30 1.6 Hypothesis & Aims 41 1.6.1 Hypothesis 41 1.6.2 Research Aims 41 1.7 Expected Outcomes 42 CHAPTER II: Materials & Methods 43 2.1 Study Design 43 2.1.1 Cohort of Study 44 2.1.2 Inheritance-based Strategy 45 1 2.1.3 Ethics for human and animals 46 2.2 Computational Methods 46 2.2.1 Pre-Processing Raw Reads 46 2.2.2 Sequencing Coverage 47 2.2.3 Variant Discovery 49 2.2.4 Annotating Variants 53 2.2.5 Evaluating Variants 55 2.3 Biological Methods 58 2.3.1 Cell Culture 58 2.3.2 Genomic DNA Sequencing 61 2.3.3 Plasmid cloning 69 2.3.4 RNA, whole exome & Whole Genome Sequencing 74 2.3.5 TOPFlash Assay 75 2.3.6 Western Blot 76 2.3.7 Morpholino Knockdown in Zebrafish 79 CHAPTER III: A mutation in COL4A2 causes autosomal dominant porencephaly with cataracts. -

Targeted Sequence Capture and Ultra High Throughput Sequencing for Gene Discovery in Inherited Diseases
Unicentre CH-1015 Lausanne http://serval.unil.ch Year : 2013 Targeted sequence capture and Ultra High Throughput sequencing for gene discovery in inherited diseases Dl GIOIA SILVIO ALESSANDRO Dl GIOIA SILVIO ALESSANDRO, 2013, Targeted sequence capture and Ultra High Throughput sequencing for gene discovery in inherited diseases Originally published at : Thesis, University of Lausanne Posted at the University of Lausanne Open Archive. http://serval.unil.ch Droits d’auteur L'Université de Lausanne attire expressément l'attention des utilisateurs sur le fait que tous les documents publiés dans l'Archive SERVAL sont protégés par le droit d'auteur, conformément à la loi fédérale sur le droit d'auteur et les droits voisins (LDA). A ce titre, il est indispensable d'obtenir le consentement préalable de l'auteur et/ou de l’éditeur avant toute utilisation d'une oeuvre ou d'une partie d'une oeuvre ne relevant pas d'une utilisation à des fins personnelles au sens de la LDA (art. 19, al. 1 lettre a). A défaut, tout contrevenant s'expose aux sanctions prévues par cette loi. Nous déclinons toute responsabilité en la matière. Copyright The University of Lausanne expressly draws the attention of users to the fact that all documents published in the SERVAL Archive are protected by copyright in accordance with federal law on copyright and similar rights (LDA). Accordingly it is indispensable to obtain prior consent from the author and/or publisher before any use of a work or part of a work for purposes other than personal use within the meaning of LDA (art. 19, para.