Qt38n028mr Nosplash A3e1d84
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Functional Roles of Bromodomain Proteins in Cancer
cancers Review Functional Roles of Bromodomain Proteins in Cancer Samuel P. Boyson 1,2, Cong Gao 3, Kathleen Quinn 2,3, Joseph Boyd 3, Hana Paculova 3 , Seth Frietze 3,4,* and Karen C. Glass 1,2,4,* 1 Department of Pharmaceutical Sciences, Albany College of Pharmacy and Health Sciences, Colchester, VT 05446, USA; [email protected] 2 Department of Pharmacology, Larner College of Medicine, University of Vermont, Burlington, VT 05405, USA; [email protected] 3 Department of Biomedical and Health Sciences, University of Vermont, Burlington, VT 05405, USA; [email protected] (C.G.); [email protected] (J.B.); [email protected] (H.P.) 4 University of Vermont Cancer Center, Burlington, VT 05405, USA * Correspondence: [email protected] (S.F.); [email protected] (K.C.G.) Simple Summary: This review provides an in depth analysis of the role of bromodomain-containing proteins in cancer development. As readers of acetylated lysine on nucleosomal histones, bromod- omain proteins are poised to activate gene expression, and often promote cancer progression. We examined changes in gene expression patterns that are observed in bromodomain-containing proteins and associated with specific cancer types. We also mapped the protein–protein interaction network for the human bromodomain-containing proteins, discuss the cellular roles of these epigenetic regu- lators as part of nine different functional groups, and identify bromodomain-specific mechanisms in cancer development. Lastly, we summarize emerging strategies to target bromodomain proteins in cancer therapy, including those that may be essential for overcoming resistance. Overall, this review provides a timely discussion of the different mechanisms of bromodomain-containing pro- Citation: Boyson, S.P.; Gao, C.; teins in cancer, and an updated assessment of their utility as a therapeutic target for a variety of Quinn, K.; Boyd, J.; Paculova, H.; cancer subtypes. -

Database Tool the Systematic Annotation of the Three Main GPCR
Database, Vol. 2010, Article ID baq018, doi:10.1093/database/baq018 ............................................................................................................................................................................................................................................................................................. Database tool The systematic annotation of the three main Downloaded from https://academic.oup.com/database/article-abstract/doi/10.1093/database/baq018/406672 by guest on 15 January 2019 GPCR families in Reactome Bijay Jassal1, Steven Jupe1, Michael Caudy2, Ewan Birney1, Lincoln Stein2, Henning Hermjakob1 and Peter D’Eustachio3,* 1European Bioinformatics Institute, Hinxton, Cambridge, CB10 1SD, UK, 2Ontario Institute for Cancer Research, Toronto, ON M5G 0A3, Canada and 3New York University School of Medicine, New York, NY 10016, USA *Corresponding author: Tel: +212 263 5779; Fax: +212 263 8166; Email: [email protected] Submitted 14 April 2010; Revised 14 June 2010; Accepted 13 July 2010 ............................................................................................................................................................................................................................................................................................. Reactome is an open-source, freely available database of human biological pathways and processes. A major goal of our work is to provide an integrated view of cellular signalling processes that spans from ligand–receptor -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

How Shelterin Protects Mammalian Telomeres 303 ANRV361-GE42-15 ARI 3 October 2008 10:10 (See 3' 5' TRF2 S Mplex
ANRV361-GE42-15 ARI 3 October 2008 10:10 ANNUAL How Shelterin Protects REVIEWS Further Click here for quick links to Annual Reviews content online, Mammalian Telomeres including: • Other articles in this volume 1 • Top cited articles Wilhelm Palm and Titia de Lange • Top downloaded articles • Our comprehensive search Laboratory for Cell Biology and Genetics, The Rockefeller University, New York, NY 10021; email: [email protected] 1Current address: Max Planck Institute of Molecular Cell Biology and Genetics, 01307 Dresden, Germany Annu. Rev. Genet. 2008. 42:301–34 Key Words First published online as a Review in Advance on ATM, ATR, cancer, NHEJ, HR August 4, 2008 by Rockefeller University on 10/13/09. For personal use only. The Annual Review of Genetics is online at Abstract genet.annualreviews.org The genomes of prokaryotes and eukaryotic organelles are usually cir- This article’s doi: cular as are most plasmids and viral genomes. In contrast, the nuclear Annu. Rev. Genet. 2008.42:301-334. Downloaded from arjournals.annualreviews.org 10.1146/annurev.genet.41.110306.130350 genomes of eukaryotes are organized on linear chromosomes, which Copyright c 2008 by Annual Reviews. require mechanisms to protect and replicate DNA ends. Eukaryotes All rights reserved navigate these problemswith the advent of telomeres, protective nucle- 0066-4197/08/1201-0301$20.00 oprotein complexes at the ends of linear chromosomes, and telomerase, the enzyme that maintains the DNA in these structures. Mammalian telomeres contain a specific protein complex, shelterin, that functions to protect chromosome ends from all aspects of the DNA damage re- sponse and regulates telomere maintenance by telomerase. -

Entrez Symbols Name Termid Termdesc 117553 Uba3,Ube1c
Entrez Symbols Name TermID TermDesc 117553 Uba3,Ube1c ubiquitin-like modifier activating enzyme 3 GO:0016881 acid-amino acid ligase activity 299002 G2e3,RGD1310263 G2/M-phase specific E3 ubiquitin ligase GO:0016881 acid-amino acid ligase activity 303614 RGD1310067,Smurf2 SMAD specific E3 ubiquitin protein ligase 2 GO:0016881 acid-amino acid ligase activity 308669 Herc2 hect domain and RLD 2 GO:0016881 acid-amino acid ligase activity 309331 Uhrf2 ubiquitin-like with PHD and ring finger domains 2 GO:0016881 acid-amino acid ligase activity 316395 Hecw2 HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2 GO:0016881 acid-amino acid ligase activity 361866 Hace1 HECT domain and ankyrin repeat containing, E3 ubiquitin protein ligase 1 GO:0016881 acid-amino acid ligase activity 117029 Ccr5,Ckr5,Cmkbr5 chemokine (C-C motif) receptor 5 GO:0003779 actin binding 117538 Waspip,Wip,Wipf1 WAS/WASL interacting protein family, member 1 GO:0003779 actin binding 117557 TM30nm,Tpm3,Tpm5 tropomyosin 3, gamma GO:0003779 actin binding 24779 MGC93554,Slc4a1 solute carrier family 4 (anion exchanger), member 1 GO:0003779 actin binding 24851 Alpha-tm,Tma2,Tmsa,Tpm1 tropomyosin 1, alpha GO:0003779 actin binding 25132 Myo5b,Myr6 myosin Vb GO:0003779 actin binding 25152 Map1a,Mtap1a microtubule-associated protein 1A GO:0003779 actin binding 25230 Add3 adducin 3 (gamma) GO:0003779 actin binding 25386 AQP-2,Aqp2,MGC156502,aquaporin-2aquaporin 2 (collecting duct) GO:0003779 actin binding 25484 MYR5,Myo1e,Myr3 myosin IE GO:0003779 actin binding 25576 14-3-3e1,MGC93547,Ywhah -

MASTER THESIS Reprogramming Human Gastrointestinal Stem Cells
MASTER THESIS Thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Engineering at the University of Applied Sciences Technikum Wien - Degree Program Tissue Engineering and Regenerative Medicine Reprogramming human gastrointestinal stem cells into insulin-secreting cells: developing a cell therapy for diabetes By: Christoph Pertl, BSc Student Number: 01306559 Supervisor 1: Qiao Joe Zhou, Ph.D. Supervisor 2: Dr. rer. nat. Elisabeth Simboeck Vienna, 15/12/2019 Declaration of Authenticity “As author and creator of this work to hand, I confirm with my signature knowledge of the relevant copyright regulations governed by higher education acts (for example see §§ 21, 42f and 57 UrhG (Austrian copyright law) as amended as well as § 14 of the Statute on Studies Act Provisions / Examination Regulations of the UAS Technikum Wien). In particular I declare that I have made use of third-party content correctly, regardless what form it may have, and I am aware of any consequences I may face on the part of the degree program director if there should be evidence of missing autonomy and independence or evidence of any intent to fraudulently achieve a pass mark for this work (see § 14 para. 1 Statute on Studies Act Provisions / Examination Regulations of the UAS Technikum Wien). I further declare that up to this date I have not published the work to hand nor have I presented it to another examination board in the same or similar form. I affirm that the version submitted matches the version in the upload tool.” Place, Date Signature 2 Kurzfassung Eine erfolgreiche Behandlung von Typ 1 Diabetes benötigt das Regenerieren verlorener insulinproduzierender Zellen und das Umgehen der Autoimmunreaktion. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Dyskerin Mutations Present in Dyskeratosis Congenita Patients Increase Oxidative Stress and DNA Damage Signalling in Dictyostelium Discoideum
cells Article Dyskerin Mutations Present in Dyskeratosis Congenita Patients Increase Oxidative Stress and DNA Damage Signalling in Dictyostelium Discoideum Javier Rodriguez-Centeno, Rosario Perona and Leandro Sastre * Instituto de Investigaciones Biomedicas, CSIC/UAM and Centro de Investigación Biomédica en Red de Enfermedades Raras, CIBERER, 28029 Madrid, Spain; [email protected] (J.R.-C.); [email protected] (R.P.) * Correspondence: [email protected]; Tel.: +3491-585-4437 Received: 11 October 2019; Accepted: 5 November 2019; Published: 8 November 2019 Abstract: Dyskerin is a protein involved in the formation of small nucleolar and small Cajal body ribonucleoproteins. These complexes participate in RNA pseudouridylation and are also components of the telomerase complex required for telomere elongation. Dyskerin mutations cause a rare disease, X-linked dyskeratosis congenita, with no curative treatment. The social amoeba Dictyostelium discoideum contains a gene coding for a dyskerin homologous protein. In this article D. discoideum mutant strains that have mutations corresponding to mutations found in dyskeratosis congenita patients are described. The phenotype of the mutant strains has been studied and no alterations were observed in pseudouridylation activity and telomere structure. Mutant strains showed increased proliferation on liquid culture but reduced growth feeding on bacteria. The results obtained indicated the existence of increased DNA damage response and reactive oxygen species, as also reported in human Dyskeratosis congenita cells and some other disease models. These data, together with the haploid character of D. discoideum vegetative cells, that resemble the genomic structure of the human dyskerin gene, located in the X chromosome, support the conclusion that D. discoideum can be a good model system for the study of this disease. -

Puromycin Selective Antibiotic for the Pac Gene Catalog # Ant-Pr-1, Ant-Pr-5 for Research Use Only Version # 13A25-MM
Puromycin Selective antibiotic for the pac gene Catalog # ant-pr-1, ant-pr-5 For research use only Version # 13A25-MM PRodUCt INFoRmAtIoN ReSIStANCe to PURomYCIN Content: The pac gene encoding a Puromycin N-acetyl-tranferase (PAC) has been isolated Puromycin hydrochloride is supplied as 1ml tubes of a 10 mg/ml solution in from a Streptomyces producing strain 1,2 . It is located in a region of the pur HEPES buffer (100% active product), filtered to sterility for customer cluster linked to the other genes d etermining the puromycin biosynthetic convenience, and cell culture tested. pathway. - ant-pr-1: 10 x 1 ml at 10 mg/ml (100 mg) The expression of pac gene confers puromycin resistance to transfected - ant-pr-5: 50 x 1 ml at 10 mg/ml (500 mg) mammalians cells 3 expressing it. In some particular conditions puromycin could also be used for selection of E. coli strains transformed with plasmids Storage and stability: carrying the pac gene. Puromycin is shipped at room temperature. Upon receipt it should be stored at 4°C or -20°C. Puromycin is stable for two years at -20°C, two years at 4°C, and 3 months at CoNdItIoNS oF SeLeCtIoN room temperature. Avoid repeated freeze-thaw cycles. Puromycin is poorly active on E. coli but is particularly useful in experiments conducted with mammalian cells. It can be used as an alternative to Quality control Neomycin system for transfection experiments. Purity controlled by HPLC: >95% Activity controlled by bioassays on bacteria and mammalian cell lines. mammalian cells The working concentrations of puromycin for mammalian cell lines range SPeCIAL hANdLING from 1 to 10 µg/ml. -
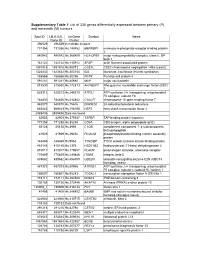
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator,