The Roles of RNA Polymerase I and III Subunits Polr1a, Polr1c, and Polr1d in Craniofacial Development BY
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
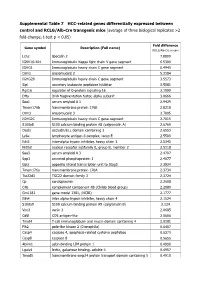
Supplemental Table 7 HCC-Related Genes Differentially Expressed Between Control and RCLG/Alb-Cre Transgenic Mice
Supplemental Table 7 HCC-related genes differentially expressed between control and RCLG/Alb-Cre transgenic mice (average of three biological replicates >2 fold-change, t-test p < 0.05) Fold difference Gene symbol Description (Full name) (RCLG/Alb-Cre vs con) Lcn2 lipocalin 2 7.8899 IGKV16-104 Immunoglobulin Kappa light chain V gene segment 6.5300 IGHG1 Immunoglobulin heavy chain C gene segment 6.4945 Orm2 orosomucoid 2 5.3184 IGHG2B Immunoglobulin heavy chain C gene segment 3.5573 Slpi secretory leukocyte peptidase inhibitor 3.5081 Rgs16 regulator of G-protein signaling 16 3.1999 Dffa DNA fragmentation factor, alpha subunit 3.0666 Saa1 serum amyloid A 1 2.9429 Tmem176b transmembrane protein 176B 2.8218 Orm3 orosomucoid 3 2.7805 IGHG2C Immunoglobulin heavy chain C gene segment 2.7015 S100a8 S100 calcium binding protein A8 (calgranulin A) 2.6769 Ocel1 occludin/ELL domain containing 1 2.6553 Ly6e lymphocyte antigen 6 complex, locus E 2.5509 Itih3 inter-alpha trypsin inhibitor, heavy chain 3 2.5345 Nr0b2 nuclear receptor subfamily 0, group B, member 2 2.5118 Saa3 serum amyloid A 3 2.4707 Spp1 secreted phosphoprotein 1 2.4077 Gats opposite strand transcription unit to Stag3 2.3934 Tmem176a transmembrane protein 176A 2.3734 Tsc22d3 TSC22 domain family 3 2.3724 Cp ceruloplasmin 2.2608 C4b complement component 4B (Childo blood group) 2.2089 Gm1381 gene model 1381, (NCBI) 2.1777 Itih4 inter alpha-trypsin inhibitor, heavy chain 4 2.1524 S100a9 S100 calcium binding protein A9 (calgranulin B) 2.124 Vnn3 vanin 3 2.0685 Cd5l CD5 antigen-like 2.0606 -

Comparative Analysis of a Teleost Skeleton Transcriptome Provides Insight Into Its Regulation
Accepted Manuscript Comparative analysis of a teleost skeleton transcriptome provides insight into its regulation Florbela A. Vieira, M.A.S. Thorne, K. Stueber, M. Darias, R. Reinhardt, M.S. Clark, E. Gisbert, D.M. Power PII: S0016-6480(13)00264-5 DOI: http://dx.doi.org/10.1016/j.ygcen.2013.05.025 Reference: YGCEN 11541 To appear in: General and Comparative Endocrinology Please cite this article as: Vieira, F.A., Thorne, M.A.S., Stueber, K., Darias, M., Reinhardt, R., Clark, M.S., Gisbert, E., Power, D.M., Comparative analysis of a teleost skeleton transcriptome provides insight into its regulation, General and Comparative Endocrinology (2013), doi: http://dx.doi.org/10.1016/j.ygcen.2013.05.025 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 Comparative analysis of a teleost skeleton transcriptome 2 provides insight into its regulation 3 4 Florbela A. Vieira1§, M. A. S. Thorne2, K. Stueber3, M. Darias4,5, R. Reinhardt3, M. 5 S. Clark2, E. Gisbert4 and D. M. Power1 6 7 1Center of Marine Sciences, Universidade do Algarve, Faro, Portugal. 8 2British Antarctic Survey – Natural Environment Research Council, High Cross, 9 Madingley Road, Cambridge, CB3 0ET, UK. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Molecular Mechanisms Regulating Mammalian Forebrain Development
Molecular Mechanisms Regulating Mammalian Forebrain Development By David Chun Cheong Tsui A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by David Chun Cheong Tsui (2013) Title: Molecular Mechanisms Regulating Mammalian Forebrain Development Name: David Chun Cheong Tsui Degree: Doctor of Philosophy, 2013 Department: Institute of Medical Sciences, University of Toronto ABSTRACT While the extrinsic factors regulating neurogenesis in the developing forebrain have been widely studied, the mechanisms downstream of the various signaling pathways are relatively ill-defined. In particular, we focused on proteins that have been implicated in cognitive dysfunction. Here, we ask what role two cell intrinsic factors play in the development of two different neurogenic compartments in the forebrain. In the first part of the thesis, the transcription factor FoxP2, which is mutated in individuals who have specific language deficits, was identified to regulate neurogenesis in the developing cortex, in part by regulating the transition from the radial precursors to the transit amplifying intermediate progenitors. Moreover, we found that ectopic expression of the human homologue of the protein promotes neurogenesis in the murine cortex, thereby acting as a gain-of-function isoform. In the second part of the thesis, the histone acetyltransferase CREB-binding protein (CBP) was identified as regulating the generation of neurons from medial ganglionic eminence precursors, similar to its role in the developing cortex. But CBP also plays a more substantial role in the expression of late interneuron markers, suggesting that it is continuously required for the various stages of neurogenesis at least in the ventral neurogenic niche. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

POLR1D Gene RNA Polymerase I and III Subunit D
POLR1D gene RNA polymerase I and III subunit D Normal Function The POLR1D gene provides instructions for making one part (subunit) of two related enzymes called RNA polymerase I and RNA polymerase III. These enzymes are involved in the production (synthesis) of ribonucleic acid (RNA), a chemical cousin of DNA. Both enzymes help synthesize a form of RNA known as ribosomal RNA (rRNA). RNA polymerase III also plays a role in the synthesis of several other forms of RNA, including transfer RNA (tRNA). Ribosomal RNA and transfer RNA assemble protein building blocks (amino acids) into functioning proteins, which is essential for the normal functioning and survival of cells. Based on its involvement in Treacher Collins syndrome, the POLR1D gene appears to play a critical role in the early development of structures that become bones and other tissues of the face. Health Conditions Related to Genetic Changes Treacher Collins syndrome At least 20 mutations in the POLR1D gene have been identified in people with Treacher Collins syndrome, a condition that affects the development of bones and other tissues of the face. These mutations appear to alter the structure and function of the POLR1D protein, which reduces the amount of functional RNA polymerase I and RNA polymerase III in cells. Consequently, less rRNA is produced. Researchers speculate that a shortage of rRNA may trigger the self-destruction (apoptosis) of certain cells involved in the early development of facial bones and tissues. The abnormal cell death could underlie the specific problems with facial development found in Treacher Collins syndrome. However, it is unclear why the effects of a reduction in rRNA are limited to facial development. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Supporting Information
Supporting Information Pratilas et al. 10.1073/pnas.0900780106 SI Text HER2) xenografts. In each of these analyses, all noncontrol Determination of Sensitivity to MEK Inhibition. Drug sensitivity probes (n ϭ 22,215) were rank-ordered according to the mag- assays were performed by using a 96-well plate and the Alamar nitude of their difference in expression after MEK inhibition Blue reagent (Invitrogen) as reported (1). The cells were treated compared with DMSO-treated controls, as measured by SAM- with PD0325901 (Pfizer) in a range of concentrations from 0.1 derived d-scores. The positions of the output genes in this signed to 500 nM (Fig. S1). ranking were then scored in a manner similar to that described by the Connectivity Map (2). This empirical and nonparametric Enrichment Analysis Over a Time Course of MEK Inhibition. Af- Kolmogorov-Smirnov-based statistic reflects an enrichment fymetrix U133A 2.0 array analysis of V600EBRAF, SkMel-28 cells, score that ranges from ϩ1toϪ1, where a score of 0 is a lack of at 0, 2, 8, and 24 h after MEK inhibition was completed as enrichment in either direction of expression, or graphically, a described (see Experimental Procedures). Robust multiarray av- random distribution of the output profile genes. Those with erage (RMA) was used to estimate the expression of probe sets. enrichment scores nearer to 1 indicated increasing correlation Each time point from2hto24wascompared with 0 h, and in between the output profile and the rank-ordered gene set from each comparison, we considered only probe sets with an expres- the experimental comparison. -

Abstracts from the 51St European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 27:870–1041 https://doi.org/10.1038/s41431-019-0408-3 MEETING ABSTRACTS Abstracts from the 51st European Society of Human Genetics Conference: Electronic Posters © European Society of Human Genetics 2019 June 16–19, 2018, Fiera Milano Congressi, Milan Italy Sponsorship: Publication of this supplement was sponsored by the European Society of Human Genetics. All content was reviewed and approved by the ESHG Scientific Programme Committee, which held full responsibility for the abstract selections. Disclosure Information: In order to help readers form their own judgments of potential bias in published abstracts, authors are asked to declare any competing financial interests. Contributions of up to EUR 10 000.- (Ten thousand Euros, or equivalent value in kind) per year per company are considered "Modest". Contributions above EUR 10 000.- per year are considered "Significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal Genetics then compared this data to de novo cases where research based PO studies were completed (N=57) in NY. E-P01.01 Results: MFSIQ (66.4) for familial deletions was Parent of origin in familial 22q11.2 deletions impacts full statistically lower (p = .01) than for de novo deletions scale intelligence quotient scores (N=399, MFSIQ=76.2). MFSIQ for children with mater- nally inherited deletions (63.7) was statistically lower D. E. McGinn1,2, M. Unolt3,4, T. B. Crowley1, B. S. Emanuel1,5, (p = .03) than for paternally inherited deletions (72.0). As E. H. Zackai1,5, E. Moss1, B. Morrow6, B. Nowakowska7,J. compared with the NY cohort where the MFSIQ for Vermeesch8, A. -

MOCHI Enables Discovery of Heterogeneous Interactome Modules in 3D Nucleome
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press MOCHI enables discovery of heterogeneous interactome modules in 3D nucleome Dechao Tian1,# , Ruochi Zhang1,# , Yang Zhang1, Xiaopeng Zhu1, and Jian Ma1,* 1Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA #These two authors contributed equally *Correspondence: [email protected] Contact To whom correspondence should be addressed: Jian Ma School of Computer Science Carnegie Mellon University 7705 Gates-Hillman Complex 5000 Forbes Avenue Pittsburgh, PA 15213 Phone: +1 (412) 268-2776 Email: [email protected] 1 Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The composition of the cell nucleus is highly heterogeneous, with different constituents forming complex interactomes. However, the global patterns of these interwoven heterogeneous interactomes remain poorly understood. Here we focus on two different interactomes, chromatin interaction network and gene regulatory network, as a proof-of-principle, to identify heterogeneous interactome modules (HIMs), each of which represents a cluster of gene loci that are in spatial contact more frequently than expected and that are regulated by the same group of transcription factors. HIM integrates transcription factor binding and 3D genome structure to reflect “transcriptional niche” in the nucleus. We develop a new algorithm MOCHI to facilitate the discovery of HIMs based on network motif clustering in heterogeneous interactomes. By applying MOCHI to five different cell types, we found that HIMs have strong spatial preference within the nucleus and exhibit distinct functional properties. Through integrative analysis, this work demonstrates the utility of MOCHI to identify HIMs, which may provide new perspectives on the interplay between transcriptional regulation and 3D genome organization. -

Regulation of Cell Fate Decisions in the Developing Forebrain by DLX
Regulation of cell fate decisions in the developing forebrain by DLX transcription factors by Mikaela Nevin A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science Medical Sciences- Medical Genetics University of Alberta © Mikaela Nevin, 2020 Abstract Introduction The Dlx1/Dlx2 double knockout (DKO) mouse exhibits major defects in forebrain development including a block in differentiation and migration of GABAergic interneurons from the subpallium to the cortex. In addition, the Dlx1/Dlx2 DKO mouse generates more oligodendrocyte progenitor cells (OPC) during forebrain development, and this occurs at the expense of interneuron generation. This phenotype suggests a role for DLX genes in mediating the neuronal-glial cell fate switch in the developing forebrain. Furthermore, expression of many oligodendroglial lineage genes is increased and occurs earlier in the DKO forebrain. As such, we hypothesized that DLX2 actively represses acquisition of oligodendroglial cell fate in a subset of forebrain neural progenitor cells by direct transcriptional repression of multiple genes required for oligodendroglial development. Here, I aimed to determine whether DLX2 represses expression of the oligodendroglial lineage genes Myt1 and Plp1 during forebrain development. In order to further characterise this gene regulatory network, I also investigated regulation of Plp1 by MYT1. I also investigated whether DLX2 may play a role in regulating progenitor cell fate in the pediatric brain tumour Diffuse Intrinsic Pontine Glioma (DIPG). Methods Chromatin immunoprecipitation with a polyclonal DLX2 antibody was performed on E13.5 mouse forebrain chromatin to determine whether DLX2 occupied candidate promoter regulatory regions of the Myt1 and Plp1 promoters, and with an MYT1 antibody on E14.5 and E18.5 forebrain to determine if MYT1 occupied candidate regulatory regions of the Plp1 promoter.