Research Article
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alphabetical Lists of the Vascular Plant Families with Their Phylogenetic
Colligo 2 (1) : 3-10 BOTANIQUE Alphabetical lists of the vascular plant families with their phylogenetic classification numbers Listes alphabétiques des familles de plantes vasculaires avec leurs numéros de classement phylogénétique FRÉDÉRIC DANET* *Mairie de Lyon, Espaces verts, Jardin botanique, Herbier, 69205 Lyon cedex 01, France - [email protected] Citation : Danet F., 2019. Alphabetical lists of the vascular plant families with their phylogenetic classification numbers. Colligo, 2(1) : 3- 10. https://perma.cc/2WFD-A2A7 KEY-WORDS Angiosperms family arrangement Summary: This paper provides, for herbarium cura- Gymnosperms Classification tors, the alphabetical lists of the recognized families Pteridophytes APG system in pteridophytes, gymnosperms and angiosperms Ferns PPG system with their phylogenetic classification numbers. Lycophytes phylogeny Herbarium MOTS-CLÉS Angiospermes rangement des familles Résumé : Cet article produit, pour les conservateurs Gymnospermes Classification d’herbier, les listes alphabétiques des familles recon- Ptéridophytes système APG nues pour les ptéridophytes, les gymnospermes et Fougères système PPG les angiospermes avec leurs numéros de classement Lycophytes phylogénie phylogénétique. Herbier Introduction These alphabetical lists have been established for the systems of A.-L de Jussieu, A.-P. de Can- The organization of herbarium collections con- dolle, Bentham & Hooker, etc. that are still used sists in arranging the specimens logically to in the management of historical herbaria find and reclassify them easily in the appro- whose original classification is voluntarily pre- priate storage units. In the vascular plant col- served. lections, commonly used methods are systema- Recent classification systems based on molecu- tic classification, alphabetical classification, or lar phylogenies have developed, and herbaria combinations of both. -

Tree Composition and Ecological Structure of Akak Forest Area
Environment and Natural Resources Research; Vol. 9, No. 4; 2019 ISSN 1927-0488 E-ISSN 1927-0496 Published by Canadian Center of Science and Education Tree Composition and Ecological Structure of Akak Forest Area Agbor James Ayamba1,2, Nkwatoh Athanasius Fuashi1, & Ayuk Elizabeth Orock1 1 Department of Environmental Science, University of Buea, Cameroon 2 Ajemalebu Self Help, Kumba, South West Region, Cameroon Correspondence: Agbor James Ayamba, Department of Environmental Science, University of Buea, Cameroon. Tel: 237-652-079-481. E-mail: [email protected] Received: August 2, 2019 Accepted: September 11, 2019 Online Published: October 12, 2019 doi:10.5539/enrr.v9n4p23 URL: https://doi.org/10.5539/enrr.v9n4p23 Abstract Tree composition and ecological structure were assessed in Akak forest area with the objective of assessing the floristic composition and the regeneration potentials. The study was carried out between April 2018 to February 2019. A total of 49 logged stumps were selected within the Akak forest spanning a period of 5 years and 20m x 20m transects were demarcated. All plants species <1cm and above were identified and recorded. Results revealed that a total of 5239 individuals from 71 families, 216 genera and 384species were identified in the study area. The maximum plants species was recorded in the year 2015 (376 species). The maximum number of species and regeneration potentials was found in the family Fabaceae, (99 species) and (31) respectively. Baphia nitida, Musanga cecropioides and Angylocalyx pynaertii were the most dominant plants specie in the years 2013, 2015 and 2017 respectively. The year 2017 depicts the highest Simpson diversity with value of (0.989) while the year 2015 show the highest Simpson dominance with value of (0.013). -

From the Late Eocene of Hordle, Southern England
Acta Palaeobotanica 59(1): 51–67, 2019 e-ISSN 2082-0259 DOI: 10.2478/acpa-2019-0006 ISSN 0001-6594 Fruit morphology, anatomy and relationships of the type species of Mastixicarpum and Eomastixia (Cornales) from the late Eocene of Hordle, southern England STEVEN R. MANCHESTER1* and MARGARET E. COLLINSON2 1 Florida Museum of Natural History, Dickinson Hall, P.O. Box 117800, Gainesville, Florida, U.S.A.; e-mail: [email protected] 2 Department of Earth Sciences, Royal Holloway University of London, Egham, Surrey TW20 0EX, United Kingdom Received 26 October 2018; accepted for publication 29 April 2019 ABSTRACT. The Mastixiaceae (Cornales) were more widespread and diverse in the Cenozoic than they are today. The fossil record includes fruits of both extant genera, Mastixia and Diplopanax, as well as several extinct genera. Two of the fossil genera, Eomastixia and Mastixicarpum, are prominent in the palaeobotanical literature, but concepts of their delimitation have varied with different authors. These genera, both based on species described 93 years ago by Marjorie Chandler from the late Eocene (Priabonian) Totland Bay Member of the Headon Hill Formation at Hordle, England, are nomenclaturally fundamental, because they were the first of a series of fos- sil mastixioid genera published from the European Cenozoic. In order to better understand the type species of Eomastixia and Mastixicarpum, we studied type specimens and topotypic material using x-ray tomography and scanning electron microscopy to supplement traditional methods of analysis, to improve our understanding of the morphology and anatomy of these fossils. Following comparisons with other fossil and modern taxa, we retain Mas- tixicarpum crassum Chandler rather than transferring it to the similar extant genus Diplopanax, and we retain Eomastixia bilocularis Chandler [=Eomastixia rugosa (Zenker) Chandler] and corroborate earlier conclusions that this species represents an extinct genus that is more closely related to Mastixia than to Diplopanax. -

Afrostyrax Lepidophyllus Extracts Exhibit in Vitro Free Radical
Moukette Moukette et al. BMC Res Notes (2015) 8:344 DOI 10.1186/s13104-015-1304-8 RESEARCH ARTICLE Open Access Afrostyrax lepidophyllus extracts exhibit in vitro free radical scavenging, antioxidant potential and protective properties against liver enzymes ion mediated oxidative damage Bruno Moukette Moukette1, Constant Anatole Pieme1*, Prosper Cabral Nya Biapa2, Vicky Jocelyne Ama Moor1, Eustace Berinyuy1 and Jeanne Yonkeu Ngogang1 Abstract Background: Several studies described the phytochemical constituents of plants in relation with the free radical scavenging property and inhibition of lipid peroxidation. This study investigated the in vitro antioxidant property, and the protective effects of ethanolic and aqueous ethanol extract of the leaves and barks of Afrostyrax lepidophyllus (Huaceae) against ion mediated oxidative damages. Methods: Four extracts (ethanol and aqueous-ethanol) from the leaves and barks of A. lepidophyllus were used in this study. The total phenols content, the antiradical and antioxidant properties were determined using standard colori- metric methods. Results: The plant extracts had a significant scavenging potential on the 2,2-diphenyl-1-picrylhydrazyl (DPPH), hydroxyl (OH), nitrite oxide (NO) and 2,2-azinobis (3-ethylbenzthiazoline)-6-sulfonic acid (ABTS) radicals with the IC50 varied between 47 and 200 µg/mL depending on the part of plant and the type of extract. The ethanol extract of A. lepidophyllus bark (GEE) showed the highest polyphenolic (35.33 0.29) and flavonoid (12.00 0.14) content. All the tested extracts demonstrated a high protective potential with the± increased of superoxide dismutase,± catalase and peroxidase activities. Conclusion: Afrostyrax lepidophyllus extracts exhibited higher antioxidant potential and significant protective poten- tial on liver enzymes. -

Systematics, Climate, and Ecology of Fossil and Extant Nyssa (Nyssaceae, Cornales) and Implications of Nyssa Grayensis Sp
East Tennessee State University Digital Commons @ East Tennessee State University Electronic Theses and Dissertations Student Works 8-2013 Systematics, Climate, and Ecology of Fossil and Extant Nyssa (Nyssaceae, Cornales) and Implications of Nyssa grayensis sp. nov. from the Gray Fossil Site, Northeast Tennessee Nathan R. Noll East Tennessee State University Follow this and additional works at: https://dc.etsu.edu/etd Part of the Biodiversity Commons, Climate Commons, Paleontology Commons, and the Plant Biology Commons Recommended Citation Noll, Nathan R., "Systematics, Climate, and Ecology of Fossil and Extant Nyssa (Nyssaceae, Cornales) and Implications of Nyssa grayensis sp. nov. from the Gray Fossil Site, Northeast Tennessee" (2013). Electronic Theses and Dissertations. Paper 1204. https://dc.etsu.edu/etd/1204 This Thesis - Open Access is brought to you for free and open access by the Student Works at Digital Commons @ East Tennessee State University. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ East Tennessee State University. For more information, please contact [email protected]. Systematics, Climate, and Ecology of Fossil and Extant Nyssa (Nyssaceae, Cornales) and Implications of Nyssa grayensis sp. nov. from the Gray Fossil Site, Northeast Tennessee ___________________________ A thesis presented to the faculty of the Department of Biological Sciences East Tennessee State University In partial fulfillment of the requirements for the degree Master of Science in Biology ___________________________ by Nathan R. Noll August 2013 ___________________________ Dr. Yu-Sheng (Christopher) Liu, Chair Dr. Tim McDowell Dr. Foster Levy Keywords: Nyssa, Endocarp, Gray Fossil Site, Miocene, Pliocene, Karst ABSTRACT Systematics, Climate, and Ecology of Fossil and Extant Nyssa (Nyssaceae, Cornales) and Implications of Nyssa grayensis sp. -
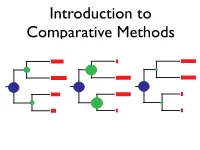
BM CC EB What Can We Learn from a Tree?
Introduction to Comparative Methods BM CC EB What can we learn from a tree? Net diversification (r) Relative extinction (ε) Peridiscaceae Peridiscaceae yllaceae yllaceae h h atop atop Proteaceae Proteaceae r r Ce Ce Tr oc T ho r M M o de c y y H H C C h r r e e a D D a o o nd o e e G G a a m m t t a a d r P r P h h e e u u c c p A p A r e a a e e a a a c a c n n a i B i B h h n d m d l m a l a m m e a e a e e t t n n c u u n n i i d i i e e e e o n o n p n p n a e a S e e S e e n n x x i i r c c a a n o n o p p h g e h g ae e l r a l r a a a a a a i i a a a e a e i b i b h y d c h d c i y i c a a c x c x c c G I a G I a n c n c c c y l y l t a a t a a e e e e e i l c i l c m l m l e c e c f a e a a f a e a a l r r l c c a i i r l e t e t a a r l a a e e u u u u o a o a a a c a c a a l a l e e e b b a a a a e e c e e c a a s c s c c e l c e l e e g e g e a a a a e e n n s e e s e e e e a a a P a P e e N N u u u S u S a e a e a a e e c c l n a l n e e a e e a e a e a e a e r a r a c c C i C i R R a e a e a e a e r c r c A A a d a a d a e i e i phanopetalaceae s r e ph s r e a a s e c s e c e e u u b a a b a e e P P r r l l e n e a a a a m m entho e e e Ha a H o a c r e c r e nt B B e p e e e c e e c a c c h e a p a a p a lo lo l l a a e s o t e s i a r a i a r r r r r a n e a n e a b a l b t a t gaceae e g e ceae a c a s c s a z e z M i a e M i a c a d e a d e ae e ae r e r a e a e a a a c c ce r e r L L i i ac a Vitaceae Vi r r C C e e ta v e v e a a c a a e ea p e ap c a c a e a P P e e l l e Ge G e e ae a t t e e p p r r ce c an u an -

Kenneth J. Wurdack 2,4 and Charles C. Davis
American Journal of Botany 96(8): 1551–1570. 2009. M ALPIGHIALES PHYLOGENETICS: GAINING GROUND ON ONE OF THE MOST RECALCITRANT CLADES IN THE ANGIOSPERM TREE OF LIFE 1 Kenneth J. Wurdack 2,4 and Charles C. Davis3,4 2 Department of Botany, Smithsonian Institution, P.O. Box 37012 NMNH MRC-166, Washington, District of Columbia 20013-7012 USA; and 3 Department of Organismic and Evolutionary Biology, Harvard University Herbaria, 22 Divinity Avenue, Cambridge, Massachusetts 02138 USA The eudicot order Malpighiales contains ~16 000 species and is the most poorly resolved large rosid clade. To clarify phyloge- netic relationships in the order, we used maximum likelihood, Bayesian, and parsimony analyses of DNA sequence data from 13 gene regions, totaling 15 604 bp, and representing all three genomic compartments (i.e., plastid: atpB , matK , ndhF, and rbcL ; mitochondrial: ccmB , cob , matR , nad1B-C , nad6, and rps3; and nuclear: 18S rDNA, PHYC, and newly developed low-copy EMB2765 ). Our sampling of 190 taxa includes representatives from all families of Malpighiales. These data provide greatly in- creased support for the recent additions of Aneulophus , Bhesa , Centroplacus , Ploiarium , and Raffl esiaceae to Malpighiales; sister relations of Phyllanthaceae + Picrodendraceae, monophyly of Hypericaceae, and polyphyly of Clusiaceae. Oxalidales + Huaceae, followed by Celastrales are successive sisters to Malpighiales. Parasitic Raffl esiaceae, which produce the world’ s largest fl owers, are confi rmed as embedded within a paraphyletic Euphorbiaceae. Novel fi ndings show a well-supported placement of Ctenolopho- naceae with Erythroxylaceae + Rhizophoraceae, sister-group relationships of Bhesa + Centroplacus , and the exclusion of Medu- sandra from Malpighiales. New taxonomic circumscriptions include the addition of Bhesa to Centroplacaceae, Medusandra to Peridiscaceae (Saxifragales), Calophyllaceae applied to Clusiaceae subfamily Kielmeyeroideae, Peraceae applied to Euphorbi- aceae subfamily Peroideae, and Huaceae included in Oxalidales. -

Systematics and Biogeography of the Clusioid Clade (Malpighiales) Brad R
Eastern Kentucky University Encompass Biological Sciences Faculty and Staff Research Biological Sciences January 2011 Systematics and Biogeography of the Clusioid Clade (Malpighiales) Brad R. Ruhfel Eastern Kentucky University, [email protected] Follow this and additional works at: http://encompass.eku.edu/bio_fsresearch Part of the Plant Biology Commons Recommended Citation Ruhfel, Brad R., "Systematics and Biogeography of the Clusioid Clade (Malpighiales)" (2011). Biological Sciences Faculty and Staff Research. Paper 3. http://encompass.eku.edu/bio_fsresearch/3 This is brought to you for free and open access by the Biological Sciences at Encompass. It has been accepted for inclusion in Biological Sciences Faculty and Staff Research by an authorized administrator of Encompass. For more information, please contact [email protected]. HARVARD UNIVERSITY Graduate School of Arts and Sciences DISSERTATION ACCEPTANCE CERTIFICATE The undersigned, appointed by the Department of Organismic and Evolutionary Biology have examined a dissertation entitled Systematics and biogeography of the clusioid clade (Malpighiales) presented by Brad R. Ruhfel candidate for the degree of Doctor of Philosophy and hereby certify that it is worthy of acceptance. Signature Typed name: Prof. Charles C. Davis Signature ( ^^^M^ *-^£<& Typed name: Profy^ndrew I^4*ooll Signature / / l^'^ i •*" Typed name: Signature Typed name Signature ^ft/V ^VC^L • Typed name: Prof. Peter Sfe^cnS* Date: 29 April 2011 Systematics and biogeography of the clusioid clade (Malpighiales) A dissertation presented by Brad R. Ruhfel to The Department of Organismic and Evolutionary Biology in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Biology Harvard University Cambridge, Massachusetts May 2011 UMI Number: 3462126 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. -

A Survey of Tricolpate (Eudicot) Phylogenetic Relationships1
American Journal of Botany 91(10): 1627±1644. 2004. A SURVEY OF TRICOLPATE (EUDICOT) PHYLOGENETIC RELATIONSHIPS1 WALTER S. JUDD2,4 AND RICHARD G. OLMSTEAD3 2Department of Botany, University of Florida, Gainesville, Florida 32611 USA; and 3Department of Biology, University of Washington, Seattle, Washington 98195 USA The phylogenetic structure of the tricolpate clade (or eudicots) is presented through a survey of their major subclades, each of which is brie¯y characterized. The tricolpate clade was ®rst recognized in 1989 and has received extensive phylogenetic study. Its major subclades, recognized at ordinal and familial ranks, are now apparent. Ordinal and many other suprafamilial clades are brie¯y diag- nosed, i.e., the putative phenotypic synapomorphies for each major clade of tricolpates are listed, and the support for the monophyly of each clade is assessed, mainly through citation of the pertinent molecular phylogenetic literature. The classi®cation of the Angiosperm Phylogeny Group (APG II) expresses the current state of our knowledge of phylogenetic relationships among tricolpates, and many of the major tricolpate clades can be diagnosed morphologically. Key words: angiosperms; eudicots; tricolpates. Angiosperms traditionally have been divided into two pri- 1992a; Chase et al., 1993; Doyle et al., 1994; Soltis et al., mary groups based on the presence of a single cotyledon 1997, 2000, 2003; KaÈllersjoÈ et al., 1998; Nandi et al., 1998; (monocotyledons, monocots) or two cotyledons (dicotyledons, Hoot et al., 1999; Savolainen et al., 2000a, b; Hilu et al., 2003; dicots). A series of additional diagnostic traits made this di- Zanis et al., 2003; Kim et al., 2004). This clade was ®rst called vision useful and has accounted for the long recognition of the tricolpates (Donoghue and Doyle, 1989), but the name these groups in ¯owering plant classi®cations. -

Relationships Within Cornales and Circumscription of Cornaceae—Matk and Rbcl Sequence Data and Effects of Outgroups and Long Branches
MOLECULAR PHYLOGENETICS AND EVOLUTION Molecular Phylogenetics and Evolution 24 (2002) 35–57 www.academicpress.com Relationships within Cornales and circumscription of Cornaceae—matK and rbcL sequence data and effects of outgroups and long branches (Jenny) Qiu-Yun Xiang,a,* Michael L. Moody,b Douglas E. Soltis,c Chaun zhu Fan,a and Pamela S. Soltis d a Department of Botany, North Carolina State University, Raleigh, NC 27695-7612, USA b Department of Ecology and Evolutionary Biology, University of Connecticut, Storrs, CT 06269-4236, USA c Department of Botany and the Genetics Institute, University of Florida, Gainesville, FL 32611-5826, USA d Florida Museum of Natural History and the Genetics Institute, University of Florida, Gainesville, FL 32611, USA Received 9 April 2001; received in revised form 1 March 2002 Abstract Phylogenetic relationships in Cornales were assessed using sequences rbcL and matK. Various combinations of outgroups were assessed for their suitability and the effects of long branches and outgroups on tree topology were examined using RASA 2.4 prior to conducting phylogenetic analyses. RASA identified several potentially problematic taxa having long branches in individual data sets that may have obscured phylogenetic signal, but when data sets were combined RASA no longer detected long branch problems. tRASA provides a more conservative measurement for phylogenetic signal than the PTP and skewness tests. The separate matK and rbcL sequence data sets were measured as the chloroplast DNA containing phylogenetic signal by RASA, but PTP and skewness tests suggested the reverse. Nonetheless, the matK and rbcL sequence data sets suggested relationships within Cornales largely congruent with those suggested by the combined matK–rbcL sequence data set that contains significant phylogenetic signal as measured by tRASA, PTP, and skewness tests. -

A Review of the Genus Curtisia (Curtisiaceae)
Bothalia 39,1: 87-96 (2009) A review of the genus Curtisia (Curtisiaceae) E. YU. YEMBATUROVA* +, B-E. VAN WYK* • and P. M. TILNEY* Keywords: anatomy, Comaceae, Curtisiaceae, Curtisia dentata (Burm.f.) C.A.Sm., revision, southern Africa ABSTRACT A review of the monotypic southern African endemic genus Curtisia Aiton is presented. Detailed studies of the fruit and seed structure provided new evidence in support of a close relationship between the family Curtisiaceae and Comaceae. Comparisons with several other members of the Comales revealed carpological similarities to certain species of Comus s.I., sometimes treated as segregate genera Dendrobenthamia Hutch, and Benthamidia Spach. We also provide information on the history of the assegai tree, Curtisia dentata (Burm.f.) C.A.Sm. and its uses, as well as a formal taxonomic revision, including nomenclature, typification, detailed description and geographical distribution. INTRODUCTION (Feder & O’Brien 1968) was applied. Suitable sections were photographed. Fruits obtained from carpological Curtisia Aiton is a monotypic genus traditionally placed collections were rehydrated and then softened by means in the family Comaceae. It is of considerable interest of prolonged heating in Strassburger mixture (water, because of the many uses of its timber and bark—but no glycerol and 96 % ethyl alcohol in equal proportions), in recent reviews of the morphology, taxonomy or anatomy accordance with traditional anatomical procedures (Pro- are available. Recent cladistic and molecular systematic zina 1960) and then sectioned either by hand or sledge studies have revealed new evidence of relationships at fam microtome. Test-reactions to identify lignification (phloro- ily level (Murrell 1993; Xiang et al. -

Mineralogy of Eocene Fossil Wood from the “Blue Forest” Locality, Southwestern Wyoming, United States
Article Mineralogy of Eocene Fossil Wood from the “Blue Forest” Locality, Southwestern Wyoming, United States George E. Mustoe 1,*, Mike Viney 2 and Jim Mills 3 1 Geology Department, Western Washington University, Bellingham, WA 98225, USA 2 College of Natural Sciences Education and Outreach Center, Colorado State University, Fort Collins, CO 80523, USA; [email protected] 3 Mills Geological, 4520 Coyote Creek Lane, Creston, CA 93432, USA; [email protected] * Correspondence: [email protected] Received: 7 December 2018; Accepted: 7 January 2019; Published: 10 January 2019 Abstract: Central Wyoming, USA, was the site of ancient Lake Gosiute during the Early Eocene. Lake Gosiute was a large body of water surrounded by subtropical forest, the lake being part of a lacustrine complex that occupied the Green River Basin. Lake level rises episodically drowned the adjacent forests, causing standing trees and fallen branches to become growth sites for algae and cyanobacteria, which encased submerged wood with thick calcareous stromatolitic coatings. The subsequent regression resulted in a desiccation of the wood, causing volume reduction, radial fractures, and localized decay. The subsequent burial of the wood in silty sediment led to a silicification of the cellular tissue. Later, chalcedony was deposited in larger spaces, as well as in the interstitial areas of the calcareous coatings. The final stage of mineralization was the precipitation of crystalline calcite in spaces that had previously remained unmineralized. The result of this multi-stage mineralization is fossil wood with striking beauty and a complex geologic origin. Keywords: fossil wood; Blue Forest; Eden Valley; Lake Gosiute; chalcedony; quartz; calcite; stromatolite; Green River Basin; Wyoming 1.