Site-Switchable Mono-O-Allylation of Polyols ✉ Hua Tang1, Yu-Biao Tian1, Hongyan Cui1, Ren-Zhe Li1, Xia Zhang 1 & Dawen Niu 1,2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Specific Determination of Airborne Sulfates and Sulfuric Acid
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1977 Specific etD ermination of Airborne Sulfates and Sulfuric Acid. Purnendu Kumar Dasgupta Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Dasgupta, Purnendu Kumar, "Specific eD termination of Airborne Sulfates and Sulfuric Acid." (1977). LSU Historical Dissertations and Theses. 3152. https://digitalcommons.lsu.edu/gradschool_disstheses/3152 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This material was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. -
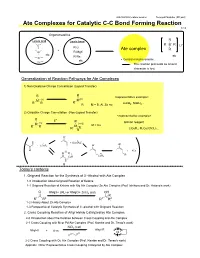
Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13
2007/09/08 Literature seminer Tomoyuki Mashiko (M1 part) Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13 Organometallics R Lewis acid Lewis base - B R B R Li+ R-Li + Ate complex R Al R-MgX etc R-Na etc Zn etc • Central metal is anionic. Cu The reaction proceeds as anionic character is lost. Generalization of Reaction Pathways for Ate Complexes 1) Non-Oxidative Charge Cancellation (Ligand Transfer) R R <representative example> M- (n) M(n) R R R LiAlH , NaBH .. R- M = B, Al, Zn etc 4 4. 2) Oxidative Charge Cancellation (Non-Ligand Transfer) <representative example> R + E R Gilman reagent M- (n) (n+2) R R M M = Cu R E LiCuR , R Cu(CN)Li .. R 2 2 2. O- Ⅱ + + [LiCuR2] - Ⅰ - O + LiCuR2 O Ⅰ O Li + RCu O R Ⅲ Ⅰ R CuR CuR2 0 Today's contents 1. Grignard Reaction for the Synthesis of 3°-Alcohol with Ate Complex 1-0 Introduction about Grignard Reaction of Ketone 1-1 Grignard Reaction of Ketone with Mg Ate Complex/ Zn Ate Complex (Prof. Ishihara and Dr. Hatano's work) O RMgX+ 2RLi or RMgCl+ ZnCl2 (cat) OH R R1 R2 R1 R2 1-2 History About Zn Ate Complex 1-3 Perspective of Catalytic Synthesis of 3°-alcohol with Grignard Reaction 2. Cross Coupling Reaction of Alkyl Halide Catalyzed by Ate Complex 2-0 Introduction about the Relation between Cross Coupling and Ate Complex 2-1 Cross Coupling with Ni or Pd Ate Complex (Prof. Kambe and Dr. Terao's work) NiCl2 (cat) Alkyl-R Alkyl-X + Ⅱ R-m Ni 2-2 Cross Coupling with Cu Ate Complex (Prof. -

Clark, Hobart, and Neu 1995
Waste Isolation Pilot Plant Compliance Certification Application Reference 135 Clark, D.L., D.E. Hobart, and M.P. Neu. 1995. Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry, Chem Revs. Vol. 95; 25-48. Submitted in accordance with 40 CFR $194.13, Submission of Reference Materials. Carera, ;., Neurcan. 3.p.. - 986 "Est~rncaonoi ?x:ier Parcrneters L'nae; Panslent anc' S:eadu Sco:z ~oi~icions,2. Unlacaness, Stc;z.:ird, and solucion ftiqo:ithrns." 9. Clark, D.L.. Floba~.D.E., Neu, M.P.. 1995 '9ctinicz Carbonate Complexes ana Thslr Irnporcccca in Ect;nide Environmencai G,~mrsny."=?em Revs. ',Jot. 05, 25-48. ON ' ; x 28.C3 398.00 Cleveland, J.M.. i 9* nGit:calRevleu of Plutonium Eov~lbrioof Environrnencal Concern. In Moueirng in Equmus S;lstms: Smrct:on, So~ubrlrtuanu tlnq of t9e Emencan Chernrcc~Societu, Pdiarnr Beacn, Fi, Series: 3521 -336. Cti~- ; x CLO.CC 220.30 . - , I. 2av1s.G.B.. Jcnnsco i 984 'Ccxxenc on Cmcamincnc Tianspor: :n fracturad PONS Media: fcr s Sjscern cS ~rciielFrcc:vres" 5y SLG~C~U,C.A., and Fmd, E.O.,' Rasc~rcesRzsecrcn, \jot. 23,i\.'o. 9. s?. : 321 - 1 322, Szpt. ., -- ? 984. 1 Qtl; I i x i 3.:: ' 48.50 , , -. - 4 Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry I David L. Clark,'~~~David E. Hobart,lb and Mary P. Neda Chemical Science and Technology Division, Los Alamas National Laboratory, Los Alamos, Mw Me& 87545, l?eThe Sciems DMm, Lawrence Berkeley Laboratory, BeBerky, California 94720, and UE G. T. Seabog imWe for Transactinium Scienae, I Livemre, California 94551 I Received May 16, 1994 (Revised Manuscript ReceM September 16, 1994) Table 1. -

Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation
No.171 No.171 ResearchResearch ArticleArticle Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation Keiichi Hirano1* and Masanobu Uchiyama1,2* 1 Graduate School of Pharmaceutical Sciences, The University of Tokyo,7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan 2 Elements Chemistry Laboratory, RIKEN,2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan E-mail: [email protected]; [email protected] Abstract: We have been focusing on development of novel synthetic methodologies based on ate complex formation. By fine-tuning of coordination environments of various main-group metals (Zn, Al, and B) as well as transition metal (Cu), a wide range of carbon-carbon bond and carbon-heteroatom bond (C–I, C–O, C–N, C–H, C–Si, and C–B) formation reactions have been realized in highly regio- and chemoselective manner taking advantage of characteristics of the elements. Keywords: Ate complex, Halogen-metal exchange, Deprotonative metalation, Chemoselectivity, Regioselectivity 1. Introduction 2. Ate Complexes Ate complexes are known to be anionic organometallic Discovery of the first mono-anion type zincate, Na[ZnEt3], complexes, whose central metals have increased valence by by James A. Wanklyn dates back to 1859,1) and even di-anion accepting Lewis basic ligands to their vacant orbitals. They type zincate, Li2[ZnMe4], was already reported by Dallas T. are attractive chemical species offering tunable reactivity due Hurd in 1948.2) Zincates experienced a long blank after these to their flexible choices of central metals, counter cations, important findings before they started to attract attentions and coordination environments. -

Effect of Substituents of Cerium Pyrazolates and Pyrrolates on Carbon Dioxide Activation
molecules Article Effect of Substituents of Cerium Pyrazolates and Pyrrolates on Carbon Dioxide Activation Uwe Bayer, Adrian Jenner, Jonas Riedmaier, Cäcilia Maichle-Mössmer and Reiner Anwander * Institute of Inorganic Chemistry, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany; [email protected] (U.B.); [email protected] (A.J.); [email protected] (J.R.); [email protected] (C.M.-M.) * Correspondence: [email protected] Abstract: Homoleptic ceric pyrazolates (pz) Ce(RR’pz)4 (R = R’ = tBu; R = R’ = Ph; R = tBu, R’ = Me) were synthesized by the protonolysis reaction of Ce[N(SiHMe2)2]4 with the corresponding pyrazole derivative. The resulting complexes were investigated in their reactivity toward CO2, revealing a significant influence of the bulkiness of the substituents on the pyrazolato ligands. The efficiency of the CO2 insertion was found to increase in the order of tBu2pz < Ph2pz < tBuMepz < Me2pz. For comparison, the pyrrole-based ate complexes [Ce2(pyr)6(m-pyr)2(thf)2][Li(thf)4]2 (pyr = pyrro- lato) and [Ce(cbz)4(thf)2][Li(thf)4] (cbz = carbazolato) were obtained via protonolysis of the cerous ate complex Ce[N(SiHMe2)2]4Li(thf) with pyrrole and carbazole, respectively. Treatment of the pyrrolate/carbazolate complexes with CO2 seemed promising, but any reversibility could not be observed. Keywords: cerium; pyrazoles; pyrroles; carbazoles; carbon dioxide Citation: Bayer, U.; Jenner, A.; Riedmaier, J.; Maichle-Mössmer, C.; Anwander, R. Effect of Substituents of 1. Introduction Cerium Pyrazolates and Pyrrolates on Rare-earth–metal complexes are capable of efficiently activating carbonylic com- Carbon Dioxide Activation. -

Exposing the Hidden Complexity of Stoichiometric and Catalytic Metathesis Reactions by Elucidation of Mg-Zn Hybrids
Exposing the hidden complexity of stoichiometric and catalytic metathesis reactions by elucidation of Mg-Zn hybrids Eva Hevia1, Jonathan Z. Chua, Pablo García-Álvarez, Alan R. Kennedy, and Matthew D. McCall WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, UK, G1 1XL Edited by Jack Halpern, University of Chicago, Chicago, IL, and approved January 14, 2010 (received for review November 17, 2009) Studying seemingly simple metathesis reactions between ZnCl2 role in these vital, new synthetic methodologies. In fact, the num- and t BuMgCl has, surprisingly, revealed a much more complex ber of organometallic compounds combining magnesium and chemistry involving mixed magnesium-zinc compounds that could zinc within the same molecule is scarce (7, 8), contrasting with be regarded as Mg-Zn hybrids. Thus, the reaction of equimolar the numerous reports on the synthesis of mixed-metal com- t amounts of ZnCl2 and BuMgCl reveals the formation of the unpre- pounds (ates) containing an alkali-metal with either magnesium μ t cedented mixed Mg-Zn complex [ðTHFÞ4Mgð -ClÞ2Znð BuÞðClÞ] (1), (magnesiates), or zinc (zincates), that show enhanced reactivity as a result of the co-complexation of the two anticipated exchange beyond the confines of the monometallic reagents from which products of the metathesis. This magnesium zincate adopts a con- they are derived (9–11). tacted ion-pair structure, closely related to Knochel’s pioneering In situ metathetical approaches are by far the most common “Turbo” Grignard reagents. Furthermore, a second coproduct iden- vehicles of choice to prepare either these mixed magnesium-zinc tified in this reaction is the solvent-separated mixed magnesium- putative intermediates or neutral organozinc reagents. -

Lithium, Sodium and Potassium Magnesiate Chemistry: a Structural
Lithium, Sodium and Potassium Magnesiate Chemistry: A Structural Overview Antonio J. Martínez-Martínez and Charles T. O’Hara WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK Until recently, deprotonative metalation reactions have been performed using organometallic compounds that contain only a single metal (e.g., organolithium reagents). Since the turn of the millennium, bimetallic compounds such as alkali metal magnesiates have begun to emerge as a new class of complementary metalating reagents. These have many benefits over traditional lithium compounds, including their enhanced stability at ambient temperatures, their tolerance of reactive functional groups and their stability in common reaction solvents. In recent years lots of attention has been focused on understanding the structure of alkali metal magnesiates in an effort to maximize synthetic efficiency and thus shed insight into approaches for future rational design. In this chapter, the diverse structural chemistry of alkali metal magnesiate compounds reported since 2007 will be summarized. KEY WORDS: Lithium, Sodium, Potassium, Magnesium, Magnesiate, X-ray Crystallography, Inverse Crowns 1.1 Introduction 1.2 Lithium magnesiate complexes 1.2.1 Alkyl/Aryl lithium magnesiate complexes 1.2.2 Amido lithium magnesiate complexes 1.2.3 Heteroleptic lithium magnesiate complexes 1.3 Sodium magnesiate complexes 1.3.1 Donor-free homo- and heteroleptic sodium magnesiate complexes 1.3.2 Introducing donors to sodium magnesiate -

A Field Method for the Determination of Tungsten in Soils
GEOLOGICAL SURVEY CIRCULAR 119 August 1951 A FIELD METHOD FOR THE DETERMINATION OF TUNGSTEN IN SOILS By F. N. Ward UNITED STATES DEPARTMENT OF THE INTERIOR Oscar L. Chapman, Secretary GEOLOGICAL SURVEY W. E. W\l"ather, Director Washington, D. C. Free on application to the Geological Survey, Washington 25, D. C. A FIELD METHOD FOR THE DETERMINATION OF TUNGSTEN IN SOILS CONTENTS Page Abstract. ___1 Discussion..... • . 3 Introduction. .. • . l References cited . • . 4 Reagents and apparatus . • . 1 Procedures. • . 2 Table 1................................. 4 ABSTRACT to meet various needs (Fernjan~H~, 1934; Fer 1yanchich, 1947; Sandell, 1946). Still other The thiocyanate-stannous chloride method for changes have been made in attempts to shorten the the determination of small quantities of tungsten method and/or obtain better precision (Geld and has been modified for use on soils in geochemical Carroll, 1949; Gentry and Sherrington, 1948). prospecting. A 0. 25 g sample of soil is fused Extraction of the tungsten thiocyanate complex ion with a modified carbonate flux in a pyrex test with an immiscible solvent results in increased tube, extracted with water, and an aliquot of the sensitivity (Sandell, 1950), and heating the re aqueous extract is treated with stannous chloride action mixture decreases the time required to .and: potassium thiocyanate in moderately concen form the complex ion (Poluektov, 1941). Accord trated hydrochloric acid. The tungsten thiocyan ingly the proposed procedure calls for a rapid ate complex ion, formed rapidly at a tempera . formation of the tungsten thiocyanate complex ion ·ture of 90C to lOOC, is extracted with a small at a temperature of 90C to 95C followed by an iso volume of isopropyl ether, and tungsten determi propyl ether extraction of the complex ion from nations are made by comparing the yellowish the cool aqueous solution. -

Principles of Chemical Recognition and Transport in Extractive
2011 Separations and Heavy Element Chemistry Research Meeting Hyatt Regency, Baltimore, MD April 26 - 29, 2011 Office of Basic Energy Sciences Chemical Sciences, Geosciences & Biosciences Division Program and Abstracts for the 2011 Separations and Heavy Element Chemistry Research Meeting Hyatt Regency Baltimore, MD April 26–29, 2011 Chemical Sciences, Geosciences, and Biosciences Division Office of Basic Energy Sciences Office of Science U.S. Department of Energy i Cover Graphics: The cover artwork is used by permission from the authors and journals for the associated covers. These covers are related to recently published papers by PIs speaking in this meeting’s program. From left to right and top to bottom they are: Christopher Cahill on p. 19 of this book. Also see C. E. Rowland and C. L. Cahill (2010) "Capturing Hydrolysis Products in the Solid State: Effects of pH on Uranyl Squarates under Ambient Conditions." Inorganic Chemistry, 49(19), 8668–8673. Wibe A. de Jong on p. 77 of this book. Also see V.A. Glezakou and W.A. de Jong, "Cluster-models for Uranyl(VI) adsorption on α-alumina." J. Phys. Chem. A 2011, 115, 1257 (2011). Jaqueline Kiplinger on p. 9 of this book. Also see Thomson, R. K.; Graves, C. R.; Scott, B. L.; Kiplinger, J. L. “Noble Reactions for the Actinides: Safe Gold-Based Access to Organouranium and Azide Complexes,” Eur. J. Inorg. Chem. 2009, 1451–1455. Jaqueline Kiplinger on p. 9 of this book. Also see Cantat, T.; Graves, C. R.; Scott, B. L.; Kiplinger, J. L. “Challenging the Metallocene Dominance in Actinide Chemistry with a Soft PNP Pincer Ligand: New Uranium Structures and Reactivity Patterns,” Angew. -

Part 1: Study Toward the Total Synthesis of Acutumine
PART 1: STUDY TOWARD THE TOTAL SYNTHESIS OF ACUTUMINE PART 2: ASYMMETRIC INTRAMOLECULAR HYDROAMINATION CATALYZED BY GROUP 3 METAL COMPLEXES by Khoi Quang Huynh A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry MONTANA STATE UNIVERSITY Bozeman, Montana October 2014 ©COPYRIGHT by Khoi Quang Huynh 2014 All Rights Reserved ii ACKNOWLEDGEMENTS I would like to express my gratitude and sincere appreciation to Professor Tom Livnghouse for his advice, assistance, and encouragement. His insight and creative thinking and enthusiasm to chemistry have inspired me. I would like to thank Professor Trevor Rainey for his generous advice and discussion. I am also grateful for the degree of independence, which he has allowed me to learn chemistry. To the members of my committee: Professor Paul Grieco, Mary Cloninger, Martin Teinzte, Stephanie Wettstein, I want to extend my appreciation for the time, effort, and valuable instructions I received in your courses. iii TABLE OF CONTENTS 1. ACUTUMINE INTRODUCTION ..................................................................................1 Background ......................................................................................................................1 Discovery .........................................................................................................................2 Biological Activity of Acutumine ....................................................................................3 Biosynthesis .....................................................................................................................4 -

Exceptionally Facile Conversion of Carboxylic Acid Salts to Aldehydes by Reductive Oxidation with Borane and Pyridinium Chlorochromate
Notes Bull. Korean Chem. Soc. 2001, Vol. 22, No. 3 325 Exceptionally Facile Conversion of Carboxylic Acid Salts to Aldehydes by Reductive Oxidation with Borane and Pyridinium Chlorochromate Jin Soon Cha,* Jae Hyung Park, and Dae Yon Lee Department of Chemistry and Institute of Natural Science, Yeungnam University, Gyongsan 712-749, Korea Received December 13, 2000 Keywords : Carboxylic acid salts, Reductive oxidation, Borane, PCC, Aldehyde synthesis. Some ten years ago, we reported that carboxylic acid form. metal salts are converted to the corresponding aldehydes in Actually carboxylate readily reacts with borane in THF to moderate to high yields by two equivalents of thexylbromo- form ate complex (acyloxyborohydride 1), the equilibrium borane-methyl sulfide1 or 9-borabicyclo[3.3.1]nonane (9- being driven over to the right by the reaction with dimethyl BBN).2 Although these procedures provided promising sulfate and hence evolution of methane gas to yield acyloxy routes to convert carboxylic acid salts to the corresponding borane 2. Finally, the oxidation of the resultant boroxine 3 aldehydes, a really clean conversion has escaped us. Thus, with PCC yields aldehydes (eq 2). the yields of aromatic aldehydes are significantly lower than those in the aliphatic series and appear to be influenced by substituents on the aromatic ring. Recently, we have found that the reductive oxidation method provides an apparently (2) ideal procedure for conversion of carboxylic acid deriva tives, such as free carboxylic acids,3 esters4 and acid chlorides5 to the corresponding aldehydes. This procedure This procedure converts both sodium and lithium salts of involves the complete reduction of such derivatives to an most aliphatic carboxylic acids to aldehydes in essentially alcohol stage with a suitable reducing agent, followed by quantitative yields, as shown in Table 1, except a,Q-unsatu- oxidation of the resultant intermediate with pyridinium chloro rated acids such as cinnamic and crotonic acids. -

The Uranato-Malate Complex
Proceedings of the Iowa Academy of Science Volume 32 Annual Issue Article 39 1925 The Uranato-Malate Complex Launcelot W. Andrews Let us know how access to this document benefits ouy Copyright ©1925 Iowa Academy of Science, Inc. Follow this and additional works at: https://scholarworks.uni.edu/pias Recommended Citation Andrews, Launcelot W. (1925) "The Uranato-Malate Complex," Proceedings of the Iowa Academy of Science, 32(1), 299-320. Available at: https://scholarworks.uni.edu/pias/vol32/iss1/39 This Research is brought to you for free and open access by the Iowa Academy of Science at UNI ScholarWorks. It has been accepted for inclusion in Proceedings of the Iowa Academy of Science by an authorized editor of UNI ScholarWorks. For more information, please contact [email protected]. Andrews: The Uranato-Malate Complex THE URANATO-MALATE COMPLEX LAUNCELOT w. ANDREWS INTRODUCTION ':rhe inirtial object of this research was the elaboration of an exact polariscopic method for the analytical determination of malic acid. The results, however, turned out to have a wider interest. A number of years ago, Walden called attention to the fact 1 that uranyl salts enhance in a remarkable degree the optical rota tion of optically active organic compounds of certain classes, par ticularly of polybasic oxyacids of the aliphatic series. This enhancement does not extend to carbohydrates in a degree suffi cient to interfere appreciably, according to observations of Gross mann2 and others. Yoder3 has laid the foundation of an analytical method based on the facts alluded to. REAGENTS Uranyl Acetate. The uranyl acetate used in this investigation was labelled "Ura nium Acetate.