Lithium, Sodium and Potassium Magnesiate Chemistry: a Structural
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Specific Determination of Airborne Sulfates and Sulfuric Acid
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1977 Specific etD ermination of Airborne Sulfates and Sulfuric Acid. Purnendu Kumar Dasgupta Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Dasgupta, Purnendu Kumar, "Specific eD termination of Airborne Sulfates and Sulfuric Acid." (1977). LSU Historical Dissertations and Theses. 3152. https://digitalcommons.lsu.edu/gradschool_disstheses/3152 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This material was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. -
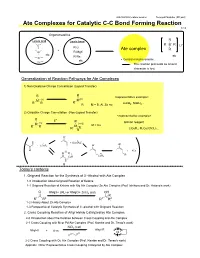
Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13
2007/09/08 Literature seminer Tomoyuki Mashiko (M1 part) Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13 Organometallics R Lewis acid Lewis base - B R B R Li+ R-Li + Ate complex R Al R-MgX etc R-Na etc Zn etc • Central metal is anionic. Cu The reaction proceeds as anionic character is lost. Generalization of Reaction Pathways for Ate Complexes 1) Non-Oxidative Charge Cancellation (Ligand Transfer) R R <representative example> M- (n) M(n) R R R LiAlH , NaBH .. R- M = B, Al, Zn etc 4 4. 2) Oxidative Charge Cancellation (Non-Ligand Transfer) <representative example> R + E R Gilman reagent M- (n) (n+2) R R M M = Cu R E LiCuR , R Cu(CN)Li .. R 2 2 2. O- Ⅱ + + [LiCuR2] - Ⅰ - O + LiCuR2 O Ⅰ O Li + RCu O R Ⅲ Ⅰ R CuR CuR2 0 Today's contents 1. Grignard Reaction for the Synthesis of 3°-Alcohol with Ate Complex 1-0 Introduction about Grignard Reaction of Ketone 1-1 Grignard Reaction of Ketone with Mg Ate Complex/ Zn Ate Complex (Prof. Ishihara and Dr. Hatano's work) O RMgX+ 2RLi or RMgCl+ ZnCl2 (cat) OH R R1 R2 R1 R2 1-2 History About Zn Ate Complex 1-3 Perspective of Catalytic Synthesis of 3°-alcohol with Grignard Reaction 2. Cross Coupling Reaction of Alkyl Halide Catalyzed by Ate Complex 2-0 Introduction about the Relation between Cross Coupling and Ate Complex 2-1 Cross Coupling with Ni or Pd Ate Complex (Prof. Kambe and Dr. Terao's work) NiCl2 (cat) Alkyl-R Alkyl-X + Ⅱ R-m Ni 2-2 Cross Coupling with Cu Ate Complex (Prof. -

Hydrogen Generation from Magnesium Hydride by Using Organic Acid Yen-Hsi Ho University of Wisconsin-Milwaukee
University of Wisconsin Milwaukee UWM Digital Commons Theses and Dissertations August 2013 Hydrogen Generation from Magnesium Hydride By Using Organic Acid Yen-Hsi Ho University of Wisconsin-Milwaukee Follow this and additional works at: https://dc.uwm.edu/etd Part of the Mechanical Engineering Commons, and the Oil, Gas, and Energy Commons Recommended Citation Ho, Yen-Hsi, "Hydrogen Generation from Magnesium Hydride By Using Organic Acid" (2013). Theses and Dissertations. 286. https://dc.uwm.edu/etd/286 This Thesis is brought to you for free and open access by UWM Digital Commons. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of UWM Digital Commons. For more information, please contact [email protected]. HYDROGEN GENERATION FROM MAGNESIUM HYDRIDE BY USING ORGANIC ACID by Yen-Hsi Ho A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Engineering at The University of Wisconsin-Milwaukee August 2013 ABSTRACT HYDROGEN GENERATION FROM MAGNESIUM HYDRIDE BY USING ORGANIC ACID by Yen-Hsi Ho The University of Wisconsin-Milwaukee, 2013 Under the Supervision of Professor Tien-Chien Jen In this paper, the hydrolysis of solid magnesium hydride has been studied with the high concentration of catalyst at the varying temperature. An organic acid (acetic acid, CH3COOH) has been chosen as the catalyst. The study has three objectives: first, using three different weights of MgH2 react with aqueous solution of acid for the hydrogen generation experiments. Secondly, utilizing acetic acid as the catalyst accelerates hydrogen generation. Third, emphasizing the combination of the three operating conditions (the weight of MgH2, the concentration of acetic acid, and the varying temperature) influence the amount of hydrogen generation. -

Clark, Hobart, and Neu 1995
Waste Isolation Pilot Plant Compliance Certification Application Reference 135 Clark, D.L., D.E. Hobart, and M.P. Neu. 1995. Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry, Chem Revs. Vol. 95; 25-48. Submitted in accordance with 40 CFR $194.13, Submission of Reference Materials. Carera, ;., Neurcan. 3.p.. - 986 "Est~rncaonoi ?x:ier Parcrneters L'nae; Panslent anc' S:eadu Sco:z ~oi~icions,2. Unlacaness, Stc;z.:ird, and solucion ftiqo:ithrns." 9. Clark, D.L.. Floba~.D.E., Neu, M.P.. 1995 '9ctinicz Carbonate Complexes ana Thslr Irnporcccca in Ect;nide Environmencai G,~mrsny."=?em Revs. ',Jot. 05, 25-48. ON ' ; x 28.C3 398.00 Cleveland, J.M.. i 9* nGit:calRevleu of Plutonium Eov~lbrioof Environrnencal Concern. In Moueirng in Equmus S;lstms: Smrct:on, So~ubrlrtuanu tlnq of t9e Emencan Chernrcc~Societu, Pdiarnr Beacn, Fi, Series: 3521 -336. Cti~- ; x CLO.CC 220.30 . - , I. 2av1s.G.B.. Jcnnsco i 984 'Ccxxenc on Cmcamincnc Tianspor: :n fracturad PONS Media: fcr s Sjscern cS ~rciielFrcc:vres" 5y SLG~C~U,C.A., and Fmd, E.O.,' Rasc~rcesRzsecrcn, \jot. 23,i\.'o. 9. s?. : 321 - 1 322, Szpt. ., -- ? 984. 1 Qtl; I i x i 3.:: ' 48.50 , , -. - 4 Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry I David L. Clark,'~~~David E. Hobart,lb and Mary P. Neda Chemical Science and Technology Division, Los Alamas National Laboratory, Los Alamos, Mw Me& 87545, l?eThe Sciems DMm, Lawrence Berkeley Laboratory, BeBerky, California 94720, and UE G. T. Seabog imWe for Transactinium Scienae, I Livemre, California 94551 I Received May 16, 1994 (Revised Manuscript ReceM September 16, 1994) Table 1. -

Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation
No.171 No.171 ResearchResearch ArticleArticle Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation Keiichi Hirano1* and Masanobu Uchiyama1,2* 1 Graduate School of Pharmaceutical Sciences, The University of Tokyo,7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan 2 Elements Chemistry Laboratory, RIKEN,2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan E-mail: [email protected]; [email protected] Abstract: We have been focusing on development of novel synthetic methodologies based on ate complex formation. By fine-tuning of coordination environments of various main-group metals (Zn, Al, and B) as well as transition metal (Cu), a wide range of carbon-carbon bond and carbon-heteroatom bond (C–I, C–O, C–N, C–H, C–Si, and C–B) formation reactions have been realized in highly regio- and chemoselective manner taking advantage of characteristics of the elements. Keywords: Ate complex, Halogen-metal exchange, Deprotonative metalation, Chemoselectivity, Regioselectivity 1. Introduction 2. Ate Complexes Ate complexes are known to be anionic organometallic Discovery of the first mono-anion type zincate, Na[ZnEt3], complexes, whose central metals have increased valence by by James A. Wanklyn dates back to 1859,1) and even di-anion accepting Lewis basic ligands to their vacant orbitals. They type zincate, Li2[ZnMe4], was already reported by Dallas T. are attractive chemical species offering tunable reactivity due Hurd in 1948.2) Zincates experienced a long blank after these to their flexible choices of central metals, counter cations, important findings before they started to attract attentions and coordination environments. -

Mechanochemical Synthesis of Hydrogen-Storage Materials Based on Aluminum, Magnesium and Their Compounds Ihor Z
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2015 Mechanochemical synthesis of hydrogen-storage materials based on aluminum, magnesium and their compounds Ihor Z. Hlova Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Materials Science and Engineering Commons, Mechanics of Materials Commons, and the Oil, Gas, and Energy Commons Recommended Citation Hlova, Ihor Z., "Mechanochemical synthesis of hydrogen-storage materials based on aluminum, magnesium and their compounds" (2015). Graduate Theses and Dissertations. 14573. https://lib.dr.iastate.edu/etd/14573 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Mechanochemical synthesis of hydrogen-storage materials based on aluminum, magnesium and their compounds by Ihor Z. Hlova A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Materials Science and Engineering Program of Study committee: Vitalij K. Pecharsky, Major Professor Karl A. Gschneidner, Jr. Marek Pruski Duane Johnson Scott Chumbley Iowa State University Ames, Iowa 2015 Copyright © Ihor Z. Hlova, 2015. All rights reserved. ii -

Hydrogen Embrittlement of Magnesium and Magnesium Alloys: a Review Mariano Kappes,A,B,∗ Mariano Iannuzzi,A,Z and Ricardo M
C168 Journal of The Electrochemical Society, 160 (4) C168-C178 (2013) 0013-4651/2013/160(4)/C168/11/$31.00 © The Electrochemical Society Hydrogen Embrittlement of Magnesium and Magnesium Alloys: A Review Mariano Kappes,a,b,∗ Mariano Iannuzzi,a,z and Ricardo M. Carranzab aNational Center for Education and Research on Corrosion and Materials Performance (NCERCAMP), The University of Akron, Akron, Ohio 44325, USA bComision´ Nacional de Energ´ıa Atomica,´ Instituto Sabato (UNSAM/CNEA), Buenos Aires 1650, Argentina Magnesium and magnesium alloys are susceptible to stress corrosion cracking in various environments, including distilled water. There is compelling evidence to conclude that SCC is assisted, at least in part, by hydrogen embrittlement. This paper reviews the thermodynamics of the Mg-H system and the kinetics of hydrogen transport. Aspects of magnesium corrosion relevant to hydrogen absorption are also discussed. Crack growth mechanisms based on delayed hydride cracking, hydrogen adsorption dislocation emission, hydrogen enhanced decohesion, and hydrogen enhanced localized plasticity have been proposed and evidence for each of them is reviewed herein. © 2013 The Electrochemical Society. [DOI: 10.1149/2.023304jes] All rights reserved. Manuscript submitted October 22, 2012; revised manuscript received January 16, 2013. Published February 26, 2013. Pure magnesium is inherently susceptible to stress corrosion crack- The nucleation and growth of MgH2 during exposure of magne- 1–3 ing (SCC) and many of its alloys suffer SCC in environments con- sium to gaseous H2 at high pressure and temperature (∼5MPaand sidered innocuous for most other engineering alloys, e.g. distilled ∼300–400◦C) has been extensively studied29–32 due to its application water.4,5 In order to prevent SCC, some authors suggest that the ap- as a solid state hydrogen storage medium. -

Electrochemical and Optical Properties of Magnesium-Alloy Hydrides Reviewed
Crystals 2012, 2, 1410-1433; doi:10.3390/cryst2041410 OPEN ACCESS crystals ISSN 2073-4352 www.mdpi.com/journal/crystals Review Electrochemical and Optical Properties of Magnesium-Alloy Hydrides Reviewed Thirugnasambandam G. Manivasagam 1,*, Kamil Kiraz 1 and Peter H. L. Notten 1,2 1 Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, MB Eindhoven 5600, The Netherlands; E-Mails: [email protected] (K.K.); [email protected] (P.H.L.N.) 2 Department of Electrical Engineering, Eindhoven University of Technology, MB Eindhoven 5600, The Netherlands * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +31-40-247-2369; Fax: +31-40-247-3481. Received: 18 April 2012; in revised form: 30 July 2012 / Accepted: 10 August 2012 / Published: 15 October 2012 Abstract: As potential hydrogen storage media, magnesium based hydrides have been systematically studied in order to improve reversibility, storage capacity, kinetics and thermodynamics. The present article deals with the electrochemical and optical properties of Mg alloy hydrides. Electrochemical hydrogenation, compared to conventional gas phase hydrogen loading, provides precise control with only moderate reaction conditions. Interestingly, the alloy composition determines the crystallographic nature of the metal- hydride: a structural change is induced from rutile to fluorite at 80 at.% of Mg in Mg-TM alloy, with ensuing improved hydrogen mobility and storage capacity. So far, 6 wt.% (equivalent to 1600 mAh/g) of reversibly stored hydrogen in MgyTM(1-y)Hx (TM: Sc, Ti) has been reported. Thin film forms of these metal-hydrides reveal interesting electrochromic properties as a function of hydrogen content. -

2020 Emergency Response Guidebook
2020 A guidebook intended for use by first responders A guidebook intended for use by first responders during the initial phase of a transportation incident during the initial phase of a transportation incident involving hazardous materials/dangerous goods involving hazardous materials/dangerous goods EMERGENCY RESPONSE GUIDEBOOK THIS DOCUMENT SHOULD NOT BE USED TO DETERMINE COMPLIANCE WITH THE HAZARDOUS MATERIALS/ DANGEROUS GOODS REGULATIONS OR 2020 TO CREATE WORKER SAFETY DOCUMENTS EMERGENCY RESPONSE FOR SPECIFIC CHEMICALS GUIDEBOOK NOT FOR SALE This document is intended for distribution free of charge to Public Safety Organizations by the US Department of Transportation and Transport Canada. This copy may not be resold by commercial distributors. https://www.phmsa.dot.gov/hazmat https://www.tc.gc.ca/TDG http://www.sct.gob.mx SHIPPING PAPERS (DOCUMENTS) 24-HOUR EMERGENCY RESPONSE TELEPHONE NUMBERS For the purpose of this guidebook, shipping documents and shipping papers are synonymous. CANADA Shipping papers provide vital information regarding the hazardous materials/dangerous goods to 1. CANUTEC initiate protective actions. A consolidated version of the information found on shipping papers may 1-888-CANUTEC (226-8832) or 613-996-6666 * be found as follows: *666 (STAR 666) cellular (in Canada only) • Road – kept in the cab of a motor vehicle • Rail – kept in possession of a crew member UNITED STATES • Aviation – kept in possession of the pilot or aircraft employees • Marine – kept in a holder on the bridge of a vessel 1. CHEMTREC 1-800-424-9300 Information provided: (in the U.S., Canada and the U.S. Virgin Islands) • 4-digit identification number, UN or NA (go to yellow pages) For calls originating elsewhere: 703-527-3887 * • Proper shipping name (go to blue pages) • Hazard class or division number of material 2. -

Effect of Substituents of Cerium Pyrazolates and Pyrrolates on Carbon Dioxide Activation
molecules Article Effect of Substituents of Cerium Pyrazolates and Pyrrolates on Carbon Dioxide Activation Uwe Bayer, Adrian Jenner, Jonas Riedmaier, Cäcilia Maichle-Mössmer and Reiner Anwander * Institute of Inorganic Chemistry, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany; [email protected] (U.B.); [email protected] (A.J.); [email protected] (J.R.); [email protected] (C.M.-M.) * Correspondence: [email protected] Abstract: Homoleptic ceric pyrazolates (pz) Ce(RR’pz)4 (R = R’ = tBu; R = R’ = Ph; R = tBu, R’ = Me) were synthesized by the protonolysis reaction of Ce[N(SiHMe2)2]4 with the corresponding pyrazole derivative. The resulting complexes were investigated in their reactivity toward CO2, revealing a significant influence of the bulkiness of the substituents on the pyrazolato ligands. The efficiency of the CO2 insertion was found to increase in the order of tBu2pz < Ph2pz < tBuMepz < Me2pz. For comparison, the pyrrole-based ate complexes [Ce2(pyr)6(m-pyr)2(thf)2][Li(thf)4]2 (pyr = pyrro- lato) and [Ce(cbz)4(thf)2][Li(thf)4] (cbz = carbazolato) were obtained via protonolysis of the cerous ate complex Ce[N(SiHMe2)2]4Li(thf) with pyrrole and carbazole, respectively. Treatment of the pyrrolate/carbazolate complexes with CO2 seemed promising, but any reversibility could not be observed. Keywords: cerium; pyrazoles; pyrroles; carbazoles; carbon dioxide Citation: Bayer, U.; Jenner, A.; Riedmaier, J.; Maichle-Mössmer, C.; Anwander, R. Effect of Substituents of 1. Introduction Cerium Pyrazolates and Pyrrolates on Rare-earth–metal complexes are capable of efficiently activating carbonylic com- Carbon Dioxide Activation. -

Magnesium Hydride for Energy Storage Applications: the Kinetics of Dehydrogenation Under Different Working Conditions
Magnesium hydride for energy storage applications: The kinetics of dehydrogenation under different working conditions Antonio Perejóna,b,*, Pedro E. Sánchez-Jiméneza, José M. Criadoa, Luis A. Pérez- Maquedaa,* aInstituto de Ciencia de Materiales de Sevilla, (C.S.I.C.-Univ. Sevilla). C. Américo Vespucio 49, Sevilla 41092. Spain bDepartamento de Química Inorgánica, Facultad de Química, Universidad de Sevilla, Sevilla 41071, Spain *Corresponding authors: [email protected] Instituto de Ciencia de Materiales de Sevilla (C.S.I.C.-Univ. Sevilla). C. Américo Vespucio 49, Sevilla 41092. Spain Tel. (+34) 95 448 95 00 Fax (+34) 95 446 01 65 [email protected] Instituto de Ciencia de Materiales de Sevilla (C.S.I.C.-Univ. Sevilla). C. Américo Vespucio 49, Sevilla 41092. Spain Tel. (+34) 95 448 95 48 Fax (+34) 95 446 01 65 1 Magnesium hydride for energy storage applications: The kinetics of dehydrogenation under different working conditions Antonio Perejóna,b,*, Pedro E. Sánchez-Jiméneza, José M. Criadoa, Luis A. Pérez- Maquedaa,* aInstituto de Ciencia de Materiales de Sevilla (C.S.I.C.-Univ. Sevilla). C. Américo Vespucio 49, Sevilla 41092. Spain bDepartamento de Química Inorgánica, Facultad de Química, Universidad de Sevilla, Sevilla 41071, Spain Abstract A new approach to the kinetics of magnesium hydride dehydrogenation is considered. A model able to predict the dehydrogenation under different experimental conditions has been proposed. A new combined kinetic analysis method, which considers the thermodynamic of the process according to the microreversibility principle, has been used for performing the kinetic analysis of data obtained under different thermal schedules at hydrogen pressures ranging from high vacuum up to 20 bar. -

Exposing the Hidden Complexity of Stoichiometric and Catalytic Metathesis Reactions by Elucidation of Mg-Zn Hybrids
Exposing the hidden complexity of stoichiometric and catalytic metathesis reactions by elucidation of Mg-Zn hybrids Eva Hevia1, Jonathan Z. Chua, Pablo García-Álvarez, Alan R. Kennedy, and Matthew D. McCall WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, UK, G1 1XL Edited by Jack Halpern, University of Chicago, Chicago, IL, and approved January 14, 2010 (received for review November 17, 2009) Studying seemingly simple metathesis reactions between ZnCl2 role in these vital, new synthetic methodologies. In fact, the num- and t BuMgCl has, surprisingly, revealed a much more complex ber of organometallic compounds combining magnesium and chemistry involving mixed magnesium-zinc compounds that could zinc within the same molecule is scarce (7, 8), contrasting with be regarded as Mg-Zn hybrids. Thus, the reaction of equimolar the numerous reports on the synthesis of mixed-metal com- t amounts of ZnCl2 and BuMgCl reveals the formation of the unpre- pounds (ates) containing an alkali-metal with either magnesium μ t cedented mixed Mg-Zn complex [ðTHFÞ4Mgð -ClÞ2Znð BuÞðClÞ] (1), (magnesiates), or zinc (zincates), that show enhanced reactivity as a result of the co-complexation of the two anticipated exchange beyond the confines of the monometallic reagents from which products of the metathesis. This magnesium zincate adopts a con- they are derived (9–11). tacted ion-pair structure, closely related to Knochel’s pioneering In situ metathetical approaches are by far the most common “Turbo” Grignard reagents. Furthermore, a second coproduct iden- vehicles of choice to prepare either these mixed magnesium-zinc tified in this reaction is the solvent-separated mixed magnesium- putative intermediates or neutral organozinc reagents.