Principles of Chemical Recognition and Transport in Extractive
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Specific Determination of Airborne Sulfates and Sulfuric Acid
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1977 Specific etD ermination of Airborne Sulfates and Sulfuric Acid. Purnendu Kumar Dasgupta Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Dasgupta, Purnendu Kumar, "Specific eD termination of Airborne Sulfates and Sulfuric Acid." (1977). LSU Historical Dissertations and Theses. 3152. https://digitalcommons.lsu.edu/gradschool_disstheses/3152 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This material was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. -

Transition-Metal-Bridged Bimetallic Clusters with Multiple Uranium–Metal Bonds
ARTICLES https://doi.org/10.1038/s41557-018-0195-4 Transition-metal-bridged bimetallic clusters with multiple uranium–metal bonds Genfeng Feng1, Mingxing Zhang1, Dong Shao 1, Xinyi Wang1, Shuao Wang2, Laurent Maron 3* and Congqing Zhu 1* Heterometallic clusters are important in catalysis and small-molecule activation because of the multimetallic synergistic effects from different metals. However, multimetallic species that contain uranium–metal bonds remain very scarce due to the difficulties in their synthesis. Here we present a straightforward strategy to construct a series of heterometallic clusters with multiple uranium–metal bonds. These complexes were created by facile reactions of a uranium precursor with Ni(COD)2 (COD, cyclooctadiene). The multimetallic clusters’ cores are supported by a heptadentate N4P3 scaffold. Theoretical investigations indicate the formation of uranium–nickel bonds in a U2Ni2 and a U2Ni3 species, but also show that they exhibit a uranium–ura- nium interaction; thus, the electronic configuration of uranium in these species is U(III)-5f26d1. This study provides further understanding of the bonding between f-block elements and transition metals, which may allow the construction of d–f hetero- metallic clusters and the investigation of their potential applications. ultimetallic molecules are of great interest because of This study offers a new opportunity to investigate d− f heteromul- their fascinating structures and multimetallic synergistic timetallic clusters with multiple uranium–metal bonds for small- Meffects for catalysis and small molecule activation1–7. Both molecule activation and catalysis. biological nitrogen fixation and industrial Haber–Bosch ammonia syntheses, for example, are thought to utilize multimetallic cata- Results and discussion lytic sites8,9. -

Inelastic Collisions of Atomic Thorium and Molecular Thorium Monoxide with Cold Helium-3
Inelastic collisions of atomic thorium and molecular thorium monoxide with cold helium-3 The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Au, Yat Shan. 2014. Inelastic collisions of atomic thorium and molecular thorium monoxide with cold helium-3. Doctoral dissertation, Harvard University. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:12274226 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Inelastic Collisions of Atomic Thorium and Molecular Thorium Monoxide with Cold Helium-3 A dissertation presented by Yat Shan Au to The Department of Physics in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Physics Harvard University Cambridge, Massachusetts November 2013 c 2013 - Yat Shan Au All rights reserved. Dissertation advisor Author Professor John Morrissey Doyle Yat Shan Au Inelastic Collisions of Atomic Thorium and Molecular Thorium Monoxide with Cold Helium-3 Abstract We measure inelastic cross sections for atomic thorium (Th) and molecular thorium monoxide (ThO) in collisions with 3He at temperatures near 1 K. We determine the 3 −17 −2 Zeeman relaxation cross section for Th ( F2) to be ∼ 2 × 10 cm at 800 mK. 3 We study electronic inelastic processes in Th ( P0) and find no quenching even after 106 collisions at 800 mK. We measure the vibrational quenching cross section for ThO (X, ν = 1) to be (7:9 ± 2:7) × 10−19 cm−2 at 800 mK. -
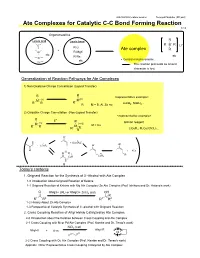
Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13
2007/09/08 Literature seminer Tomoyuki Mashiko (M1 part) Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13 Organometallics R Lewis acid Lewis base - B R B R Li+ R-Li + Ate complex R Al R-MgX etc R-Na etc Zn etc • Central metal is anionic. Cu The reaction proceeds as anionic character is lost. Generalization of Reaction Pathways for Ate Complexes 1) Non-Oxidative Charge Cancellation (Ligand Transfer) R R <representative example> M- (n) M(n) R R R LiAlH , NaBH .. R- M = B, Al, Zn etc 4 4. 2) Oxidative Charge Cancellation (Non-Ligand Transfer) <representative example> R + E R Gilman reagent M- (n) (n+2) R R M M = Cu R E LiCuR , R Cu(CN)Li .. R 2 2 2. O- Ⅱ + + [LiCuR2] - Ⅰ - O + LiCuR2 O Ⅰ O Li + RCu O R Ⅲ Ⅰ R CuR CuR2 0 Today's contents 1. Grignard Reaction for the Synthesis of 3°-Alcohol with Ate Complex 1-0 Introduction about Grignard Reaction of Ketone 1-1 Grignard Reaction of Ketone with Mg Ate Complex/ Zn Ate Complex (Prof. Ishihara and Dr. Hatano's work) O RMgX+ 2RLi or RMgCl+ ZnCl2 (cat) OH R R1 R2 R1 R2 1-2 History About Zn Ate Complex 1-3 Perspective of Catalytic Synthesis of 3°-alcohol with Grignard Reaction 2. Cross Coupling Reaction of Alkyl Halide Catalyzed by Ate Complex 2-0 Introduction about the Relation between Cross Coupling and Ate Complex 2-1 Cross Coupling with Ni or Pd Ate Complex (Prof. Kambe and Dr. Terao's work) NiCl2 (cat) Alkyl-R Alkyl-X + Ⅱ R-m Ni 2-2 Cross Coupling with Cu Ate Complex (Prof. -

Catalytic Organic Transformations Mediated by Actinide Complexes
Inorganics 2015, 3, 392-428; doi:10.3390/inorganics3040392 OPEN ACCESS inorganics ISSN 2304-6740 www.mdpi.com/journal/inorganics Review Catalytic Organic Transformations Mediated by Actinide Complexes Isabell S. R. Karmel, Rami J. Batrice and Moris S. Eisen * Schulich Faculty of Chemistry, Technion—Israel Institute of Technology, Technion City, Haifa 32000, Israel; E-Mails: [email protected] (I.S.R.K.); [email protected] (R.J.B.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel./Fax: +972-4-829-2680. Academic Editors: Stephen Mansell and Steve Liddle Received: 16 September 2015 / Accepted: 9 October 2015 / Published: 30 October 2015 Abstract: This review article presents the development of organoactinides and actinide coordination complexes as catalysts for homogeneous organic transformations. This chapter introduces the basic principles of actinide catalysis and deals with the historic development of actinide complexes in catalytic processes. The application of organoactinides in homogeneous catalysis is exemplified in the hydroelementation reactions, such as the hydroamination, hydrosilylation, hydroalkoxylation and hydrothiolation of alkynes. Additionally, the use of actinide coordination complexes for the catalytic polymerization of α-olefins and the ring opening polymerization of cyclic esters is presented. The last part of this review article highlights novel catalytic transformations mediated by actinide compounds and gives an outlook to the further potential of this field. Keywords: organoactinides; actinide coordination complexes; homogeneous catalysis; hydroelementations; polymerization of olefins; ROP; activation of heterocumulenes 1. Introduction The beginning of modern organoactinide chemistry is often attributed to the synthesis of 8 uranocene, [(η -C8H8)2U] in 1968, as the analogous compound to ferrocene and other transition metal metallocenes [1,2]. -

Clark, Hobart, and Neu 1995
Waste Isolation Pilot Plant Compliance Certification Application Reference 135 Clark, D.L., D.E. Hobart, and M.P. Neu. 1995. Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry, Chem Revs. Vol. 95; 25-48. Submitted in accordance with 40 CFR $194.13, Submission of Reference Materials. Carera, ;., Neurcan. 3.p.. - 986 "Est~rncaonoi ?x:ier Parcrneters L'nae; Panslent anc' S:eadu Sco:z ~oi~icions,2. Unlacaness, Stc;z.:ird, and solucion ftiqo:ithrns." 9. Clark, D.L.. Floba~.D.E., Neu, M.P.. 1995 '9ctinicz Carbonate Complexes ana Thslr Irnporcccca in Ect;nide Environmencai G,~mrsny."=?em Revs. ',Jot. 05, 25-48. ON ' ; x 28.C3 398.00 Cleveland, J.M.. i 9* nGit:calRevleu of Plutonium Eov~lbrioof Environrnencal Concern. In Moueirng in Equmus S;lstms: Smrct:on, So~ubrlrtuanu tlnq of t9e Emencan Chernrcc~Societu, Pdiarnr Beacn, Fi, Series: 3521 -336. Cti~- ; x CLO.CC 220.30 . - , I. 2av1s.G.B.. Jcnnsco i 984 'Ccxxenc on Cmcamincnc Tianspor: :n fracturad PONS Media: fcr s Sjscern cS ~rciielFrcc:vres" 5y SLG~C~U,C.A., and Fmd, E.O.,' Rasc~rcesRzsecrcn, \jot. 23,i\.'o. 9. s?. : 321 - 1 322, Szpt. ., -- ? 984. 1 Qtl; I i x i 3.:: ' 48.50 , , -. - 4 Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry I David L. Clark,'~~~David E. Hobart,lb and Mary P. Neda Chemical Science and Technology Division, Los Alamas National Laboratory, Los Alamos, Mw Me& 87545, l?eThe Sciems DMm, Lawrence Berkeley Laboratory, BeBerky, California 94720, and UE G. T. Seabog imWe for Transactinium Scienae, I Livemre, California 94551 I Received May 16, 1994 (Revised Manuscript ReceM September 16, 1994) Table 1. -

Diphosphazide-Supported Trialkyl Thorium(IV) Complex Tara K
pubs.acs.org/Organometallics Communication Diphosphazide-Supported Trialkyl Thorium(IV) Complex Tara K. K. Dickie, Ashraf A. Aborawi, and Paul G. Hayes* Cite This: Organometallics 2020, 39, 2047−2052 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: The potassium salt of a new ligand, KLP=N3 (2, κ5 i i − LP=N3 = -2,5-[(4- PrC6H4)N3 P Pr2]2N(C4H2) ), that features two units of the rare phosphazide (RN3 PR3) functionality was synthesized via an incomplete Staudinger reaction between K[2,5- i i ( Pr2P)2N(C4H2)] (1)and4-PrC6H4N3. The diphosphazide ligand was transferred to a thorium(IV) metal center using salt metathesis strategies, yielding LP=N3ThCl3 (3), which contains two intact and coordinated phosphazides. Reaction of complex 3 with 3 equiv of LiCH2SiMe3 resulted in formation of the trialkyl thorium species LP=N3Th(CH2SiMe3)3 (4). In contrast, attempts to synthesize an organothorium complex supported by the previously κ3 reported bisphosphinimine ligand LP=N (LP=N = -2,5- i i − ff [(4- PrC6H4)N P Pr2]2N(C4H2) )aorded the cyclometalated * * κ4 i i i i 2− dialkyl complex L P=NTh(CH2SiMe3)2 (6,L PN = -2-[(4- PrC6H3)N P Pr2]-5-[(4- PrC6H4)N P Pr2]N(C4H2) ) and 1 equiv of free tetramethylsilane. 1 he Staudinger reaction, discovered in 1919, introduced the facile loss of N2, and, accordingly, were overlooked as ′ ’ T the formation of a phosphinimine group (R3P NR ) via viable functional groups in ligand design. Since Staudinger s the reaction of a tertiary phosphine (R3P) with an organic original work, multiple methods have been developed to ′ azide (N3R ), resulting in concomitant loss of N2. -

Extraction of Thorium Oxide from Monazite Ore
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Supervised Undergraduate Student Research Chancellor’s Honors Program Projects and Creative Work 5-2019 Extraction of Thorium Oxide from Monazite Ore Makalee Ruch University of Tennessee, Knoxville, [email protected] Chloe Frame University of Tennessee, Knoxville Molly Landon University of Tennessee, Knoxville Ralph Laurel University of Tennessee, Knoxville Annabelle Large University of Tennessee, Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_chanhonoproj Part of the Environmental Chemistry Commons, Geological Engineering Commons, and the Other Chemical Engineering Commons Recommended Citation Ruch, Makalee; Frame, Chloe; Landon, Molly; Laurel, Ralph; and Large, Annabelle, "Extraction of Thorium Oxide from Monazite Ore" (2019). Chancellor’s Honors Program Projects. https://trace.tennessee.edu/utk_chanhonoproj/2294 This Dissertation/Thesis is brought to you for free and open access by the Supervised Undergraduate Student Research and Creative Work at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Chancellor’s Honors Program Projects by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Extraction of Thorium Oxide from Monazite Ore Dr. Robert Counce Department of Chemical and Biomolecular Engineering University of Tennessee Chloe Frame Molly Landon Annabel Large Ralph Laurel Makalee Ruch CBE 488: Honors -

Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation
No.171 No.171 ResearchResearch ArticleArticle Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation Keiichi Hirano1* and Masanobu Uchiyama1,2* 1 Graduate School of Pharmaceutical Sciences, The University of Tokyo,7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan 2 Elements Chemistry Laboratory, RIKEN,2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan E-mail: [email protected]; [email protected] Abstract: We have been focusing on development of novel synthetic methodologies based on ate complex formation. By fine-tuning of coordination environments of various main-group metals (Zn, Al, and B) as well as transition metal (Cu), a wide range of carbon-carbon bond and carbon-heteroatom bond (C–I, C–O, C–N, C–H, C–Si, and C–B) formation reactions have been realized in highly regio- and chemoselective manner taking advantage of characteristics of the elements. Keywords: Ate complex, Halogen-metal exchange, Deprotonative metalation, Chemoselectivity, Regioselectivity 1. Introduction 2. Ate Complexes Ate complexes are known to be anionic organometallic Discovery of the first mono-anion type zincate, Na[ZnEt3], complexes, whose central metals have increased valence by by James A. Wanklyn dates back to 1859,1) and even di-anion accepting Lewis basic ligands to their vacant orbitals. They type zincate, Li2[ZnMe4], was already reported by Dallas T. are attractive chemical species offering tunable reactivity due Hurd in 1948.2) Zincates experienced a long blank after these to their flexible choices of central metals, counter cations, important findings before they started to attract attentions and coordination environments. -

Summaries of FY 1997 Research in the Chemical Sciences
DOE/NBM-1098 Rev.-1 September 1997 T O EN FE TM N R E A R P G E Y D U • • A N C I I T R E D E M ST A ATES OF Summaries of FY 1997 Research in the Chemical Sciences U.S. Department of Energy Office of Energy Research Division of Chemical Sciences A searchable version of this summary book is available at the following web address: http://websrv.er.doe.gov/asp/search.asp This search tool is also accessible from the Chemical Sciences web page at: http://www.er.doe.gov/production/bes/chm/chmhome.html Available to DOE and DOE contractors from the Office of Scientific and Technical Information, P.O. Box 62, Oak Ridge, TN 37831; prices available from (423) 576-8401 Available to the public from the U.S. Department of Commerce, Technology Administration, National Technical Information Service, Springfield, VA 22161 This document was produced under contract number DE-AC05-76OR00033 between the U.S. Department of Energy and Oak Ridge Associated Universities. ORISE 97-1555 CONTENTS CONTENTS PREFACE ........................................................................ vii Oak Ridge National Laboratory.............................. 42 DIVISION OF CHEMICAL SCIENCES ..................... viii Pacific Northwest National Laboratory .................. 44 PROGRAM DESCRIPTIONS ........................................ ix Heavy Element Chemistry ....................................... 45 LABORATORY ADMINISTRATION ......................... xiii Argonne National Laboratory ................................. 45 Lawrence Berkeley National Laboratory............... -

Process for the Production of Uranium Trifluoride
United States Patent im [in 3,964,965 Tagawa [45] June 24, 1976 [54] PROCESS FOR THE PRODUCTION OF URANIUM TRIFLUORIDE [56] References Cited [75] Inventor: Hiroaki Tagawa, Tokaimura, Japan UNITED STATES PATENTS [73] Assignee: Japan Atomic Energy Research 3,034,855 5/1962 Jenkins et al 423/258 Institute, Tokyo, Japan [22] Filed: Dec. 20, 1973 Primary Examiner—Stephen J. Lechert, Jr. Attorney, Agent, or Firm—Stevens, Davis, Miller & [21] Appl. No.: 426,593 Mosher [30] Foreign Application Priority Data [57] ABSTRACT Dec. 26, 1972 Japan 47-129560 A novel method is disclosed for producing a pure ura- nium trifluoride efficiently. Said method is character- [52] U.S. CI 423/258; 423/259; ized by heating a mixture of uranium tetrafluoride and 252/301.1 R uranium nitride in an inert gas stream or under [51] Int. CI.2. C01G 43/06 vacuum. [58] Field of Search 423/258, 259; 252/301.1 R 2 Claims, No Drawings 3,976, 1 2 PROCESS FOR THE PRODUCTION OF URANIUM DETAILED DESCRIPTION OF INVENTION TRIFLUORIDE According to the present invention, uranium trifluo- ride is produced by heating a mixture of uranium tetra- BACKGROUND OF THE INVENTION 5 fluoride and uranium nitride in the form of powder or 1. Field of the Invention molding in a stream of inert gas or under vacuum. In The present invention relates to a method for pro- this invention, uranium sesquinitride (U2N3) or ura- duction of pure uranium trifluoride characterized by nium mononitride (UN) can be used for the starting heating a mixture of uranium tetrafluoride and uranium material. -

Alkaline-Earth Metal Compounds Oddities and Applications 45 Topics in Organometallic Chemistry
Topics in Organometallic Chemistry 45 Sjoerd Harder Editor Alkaline-Earth Metal Compounds Oddities and Applications 45 Topics in Organometallic Chemistry Editorial Board: M. Beller l J. M. Brown l P. H. Dixneuf A. Fu¨rstner l L. J. Gooßen P. Hofmann l T. Ikariya l S. Nolan L. A. Oro l Q.-L. Zhou Topics in Organometallic Chemistry Recently Published Volumes Inventing Reactions Iridium Catalysis Volume Editor: Lukas J. Gooßen Volume Editor: P. G. Andersson Vol. 44, 2013 Vol. 34, 2011 Hydrofunctionalization Iron Catalysis – Fundamentals and Volume Editors: Valentine P. Ananikov, Applications Masato Tanaka Volume Editor: B. Plietker Vol. 43, 2013 Vol. 33, 2011 Organometallics as Catalysts Medicinal Organometallic Chemistry in the Fine Chemical Industry Volume Editors: G. Jaouen, N. Metzler-Nolte Volume Editors: Matthias Beller, Vol. 32, 2010 Hans-Ulrich Blaser C-X Bond Formation Vol. 42, 2012 Volume Editor: A. Vigalok Modern Organoaluminum Reagents: Vol. 31, 2010 Preparation, Structure, Reactivity and Use Transition Metal Complexes of Neutral Volume Editors: Simon Woodward, h1-Carbon Ligands Samuel Dagorne Volume Editors: R. Chauvin, Y. Canac Vol. 41, 2012 Vol. 30, 2010 Organometallic Pincer Chemistry Photophysics of Organometallics Volume Editors: Gerard van Koten, Volume Editor: A. J. Lees David Milstein Vol. 29, 2010 Vol. 40, 2012 Molecular Organometallic Materials Organometallics and Renewables for Optics Volume Editors: Michael A. R. Meier, Volume Editors: H. Le Bozec, V. Guerchais Bert M. Weckhuysen, Pieter C. A. Bruijnincx Vol. 28, 2010 Vol. 39, 2012 Conducting and Magnetic Organometallic Transition Metal Catalyzed Enantioselective Molecular Materials Allylic Substitution in Organic Synthesis Volume Editors: M. Fourmigue´, L. Ouahab Volume Editor: Uli Kazmaier Vol.