Synthesis of Azulenes Via the -Halo Diazo Ketone Strategy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Synthesis of Molecules Containing Quaternary Stereogenic Centers Via the Intramolecular Asymmetric Heck Reaction
The Synthesis of Molecules Containing Quaternary Stereogenic Centers via the Intramolecular Asymmetric Heck Reaction Reported by Eric P. Gillis April 19, 2007 INTRODUCTION The enantioselective synthesis of compounds containing quaternary stereocenters continues to be a challenging endeavor and is a field of intense interest and development.1,,,,2 3 4 5 From the perspective of methods development, the asymmetric synthesis of compounds containing quaternary stereocenters poses a distinct challenge due to the sterically congested environment in which C-C bonds must be formed and stereochemical information must be transferred. Existing asymmetric methods have been applied successfully in desymmetrization processes that transform prochiral quaternary centers into stereodefined quaternary centers. However, for processes in which a quaternary stereocenter is created directly via a C-C bond forming reaction, only a few asymmetric reactions have been fruitful. The diversity in the array of methods that exist for the synthesis of compounds containing quaternary stereocenters is necessitated in part because of the number of distinct quaternary stereocenter architectures that are possible (Scheme 1). The construction of acyclic quaternary stereocenters is particularly challenging due to the number of degrees of freedom associated with these structures. However, promising results for the construction of these stereocenters via allylation,6 allylic alklyation,7 and Michael addition8 have been reported recently. For the synthesis of cyclic quaternary stereocenters a number of methods have been employed successfully including the intramolecular Heck reaction,9 alkylation,10 arylation,11 and Michael addition12. Fused bicyclic quaternary stereocenters are generally accessed via the desymmetrization of a prochiral quaternary center. In this regard, theoretically any SCHEME 1. -

Specific Determination of Airborne Sulfates and Sulfuric Acid
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1977 Specific etD ermination of Airborne Sulfates and Sulfuric Acid. Purnendu Kumar Dasgupta Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Dasgupta, Purnendu Kumar, "Specific eD termination of Airborne Sulfates and Sulfuric Acid." (1977). LSU Historical Dissertations and Theses. 3152. https://digitalcommons.lsu.edu/gradschool_disstheses/3152 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This material was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. -

Expanding the Scope of Thiophene Based Semiconductors: Perfluoroalkylated Materials and Fused Thienoacenes
Expanding the Scope of Thiophene Based Semiconductors: Perfluoroalkylated Materials and Fused Thienoacenes Hayden Thompson Black A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry Chapel Hill 2012 Approved By: Dr. Valerie Sheares Ashby Dr. James Cahoon Dr. Carrie Donley Dr. Wei You Dr. Malcolm Forbes ABSTRACT HAYDEN THOMPSON BLACK: Expanding the Scope of Thiophene Based Semiconductors: Perfluoroalkylated Materials and Fused thienoacenes (Under the direction of Valerie Sheares Ashby) Thiophene based semiconductors with new molecular and macromolecular structures were explored for applications in field effect transistors. Perfluoroalkylation was studied both as a means for controlling the self-assembly properties of polythiophenes, as well as modifying the molecular orbital energies of a series of oligothiophenes. End-perfluoroalkylation of poly(3-hexylthiophene) resulted in interesting self-assembly of the polymer into a bilayer vesicle. Similar fluorophilic assembly may be useful for controlling blend morphologies in heterojunction based devices. On the other hand, perfluoroalkylation of small molecule thiophene semiconductors leads to low lying LUMO levels, and can be used to promote electron injection for n-type transistor devices. This strategy was employed in combination with a π-electron deficient benzothiadiazole to afford a new n-type semiconductor with an exceptionally low LUMO. Monoperfluoroalkylated oligothiophenes were also synthesized and studied in field effect transistors for the first time. In addition, two new fused thienoacene compounds were synthesized and their crystal structures were analyzed. The fused compounds showed exceptional π-π stacking and assembled into well defined one-dimensional microcrystals from the vapor phase. -

Thiophene-Based Aldehyde Derivatives for Functionalizable
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archive Ouverte en Sciences de l'Information et de la Communication Thiophene-based aldehyde derivatives for functionalizable & adhesive semiconducting polymers Emin Istif, Daniele Mantione, Lorenzo Vallan, Georges Hadziioannou, Cyril Brochon, Eric Cloutet, Eleni Pavlopoulou To cite this version: Emin Istif, Daniele Mantione, Lorenzo Vallan, Georges Hadziioannou, Cyril Brochon, et al.. Thiophene-based aldehyde derivatives for functionalizable & adhesive semiconducting polymers. ACS Applied Materials & Interfaces, Washington, D.C. : American Chemical Society, 2020, 10.1021/ac- sami.9b21058. hal-02467037 HAL Id: hal-02467037 https://hal.archives-ouvertes.fr/hal-02467037 Submitted on 4 Feb 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Thiophene-based aldehyde derivatives for functionalizable & adhesive semiconducting polymers Emin Istif,† Daniele Mantione,†* Lorenzo Vallan,† Georges Hadziioannou,† Cyril Brochon,† Eric Cloutet,†* and Eleni Pavlopoulou†* †Laboratoire de Chimie des Polymères Organiques (LCPO - UMR 5629), Bordeaux INP, Université de Bordeaux, CNRS, 16 Av. Pey-Berland, 33607, Pessac, France. Keywords EDOT-Aldehyde, thiophene-Aldehyde, PEDOT, conductive polymers, adhesion, electrode materials Abstract The pursuit for novelty in the field of (bio)electronics demands for new and better performing (semi)conductive materials. -
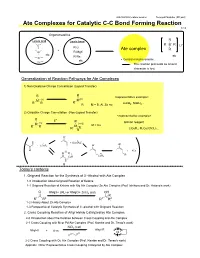
Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13
2007/09/08 Literature seminer Tomoyuki Mashiko (M1 part) Ate Complexes for Catalytic C-C Bond Forming Reaction 1/13 Organometallics R Lewis acid Lewis base - B R B R Li+ R-Li + Ate complex R Al R-MgX etc R-Na etc Zn etc • Central metal is anionic. Cu The reaction proceeds as anionic character is lost. Generalization of Reaction Pathways for Ate Complexes 1) Non-Oxidative Charge Cancellation (Ligand Transfer) R R <representative example> M- (n) M(n) R R R LiAlH , NaBH .. R- M = B, Al, Zn etc 4 4. 2) Oxidative Charge Cancellation (Non-Ligand Transfer) <representative example> R + E R Gilman reagent M- (n) (n+2) R R M M = Cu R E LiCuR , R Cu(CN)Li .. R 2 2 2. O- Ⅱ + + [LiCuR2] - Ⅰ - O + LiCuR2 O Ⅰ O Li + RCu O R Ⅲ Ⅰ R CuR CuR2 0 Today's contents 1. Grignard Reaction for the Synthesis of 3°-Alcohol with Ate Complex 1-0 Introduction about Grignard Reaction of Ketone 1-1 Grignard Reaction of Ketone with Mg Ate Complex/ Zn Ate Complex (Prof. Ishihara and Dr. Hatano's work) O RMgX+ 2RLi or RMgCl+ ZnCl2 (cat) OH R R1 R2 R1 R2 1-2 History About Zn Ate Complex 1-3 Perspective of Catalytic Synthesis of 3°-alcohol with Grignard Reaction 2. Cross Coupling Reaction of Alkyl Halide Catalyzed by Ate Complex 2-0 Introduction about the Relation between Cross Coupling and Ate Complex 2-1 Cross Coupling with Ni or Pd Ate Complex (Prof. Kambe and Dr. Terao's work) NiCl2 (cat) Alkyl-R Alkyl-X + Ⅱ R-m Ni 2-2 Cross Coupling with Cu Ate Complex (Prof. -
 in THF](https://docslib.b-cdn.net/cover/3589/hydrogenationn-of-4-octyne-catalyzedd-by-pd-m-w-cf3-2c6h3-bian-ma-in-thf-533589.webp)
Hydrogenationn of 4-Octyne Catalyzedd by Pd[(M^W'- (CF3)2C6H3)) Bian](Ma) in THF
UvA-DARE (Digital Academic Repository) Palladium-catalyzed stereoselective hydrogenation of alkynes to (Z)-alkenes in common solvents and supercritical CO2 Kluwer, A.M. Publication date 2004 Link to publication Citation for published version (APA): Kluwer, A. M. (2004). Palladium-catalyzed stereoselective hydrogenation of alkynes to (Z)- alkenes in common solvents and supercritical CO2. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:04 Oct 2021 5 Kineti5 cc study and Spectroscopic Investigationn of the semi- hydrogenationn of 4-octyne catalyzedd by Pd[(m^w'- (CF3)2C6H3)) bian](ma) in THF 5.11 Introduction Homogeneouss hydrogenation by transition metal complexes has played a key role in the fundamental understandingg of catalytic reactions and has proven to be of great utility in practical applications. -

Clark, Hobart, and Neu 1995
Waste Isolation Pilot Plant Compliance Certification Application Reference 135 Clark, D.L., D.E. Hobart, and M.P. Neu. 1995. Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry, Chem Revs. Vol. 95; 25-48. Submitted in accordance with 40 CFR $194.13, Submission of Reference Materials. Carera, ;., Neurcan. 3.p.. - 986 "Est~rncaonoi ?x:ier Parcrneters L'nae; Panslent anc' S:eadu Sco:z ~oi~icions,2. Unlacaness, Stc;z.:ird, and solucion ftiqo:ithrns." 9. Clark, D.L.. Floba~.D.E., Neu, M.P.. 1995 '9ctinicz Carbonate Complexes ana Thslr Irnporcccca in Ect;nide Environmencai G,~mrsny."=?em Revs. ',Jot. 05, 25-48. ON ' ; x 28.C3 398.00 Cleveland, J.M.. i 9* nGit:calRevleu of Plutonium Eov~lbrioof Environrnencal Concern. In Moueirng in Equmus S;lstms: Smrct:on, So~ubrlrtuanu tlnq of t9e Emencan Chernrcc~Societu, Pdiarnr Beacn, Fi, Series: 3521 -336. Cti~- ; x CLO.CC 220.30 . - , I. 2av1s.G.B.. Jcnnsco i 984 'Ccxxenc on Cmcamincnc Tianspor: :n fracturad PONS Media: fcr s Sjscern cS ~rciielFrcc:vres" 5y SLG~C~U,C.A., and Fmd, E.O.,' Rasc~rcesRzsecrcn, \jot. 23,i\.'o. 9. s?. : 321 - 1 322, Szpt. ., -- ? 984. 1 Qtl; I i x i 3.:: ' 48.50 , , -. - 4 Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry I David L. Clark,'~~~David E. Hobart,lb and Mary P. Neda Chemical Science and Technology Division, Los Alamas National Laboratory, Los Alamos, Mw Me& 87545, l?eThe Sciems DMm, Lawrence Berkeley Laboratory, BeBerky, California 94720, and UE G. T. Seabog imWe for Transactinium Scienae, I Livemre, California 94551 I Received May 16, 1994 (Revised Manuscript ReceM September 16, 1994) Table 1. -

Supporting Information
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is © The Royal Society of Chemistry 2014 Supplementary Material Nickel-Catalyzed Substitution Reactions of Propargyl Halides with Organotitanium Reagents Qing-Han Li,a,b,* Jung-Wei Liao,a Yi-Ling Huang,a Ruei-Tang Chianga and Han-Mou Gaua,* a Department of Chemistry, National Chung Hsing University, Taichung 402, Taiwan b College of Chemistry and Environmental Protection Engineering, Southwest University for Nationalities, Chengdu 610041, China e-mail: [email protected] - S1- Table of Contents I. 1H and 13C NMR Spectra of Aryltitanium Reagents S3 1. (2-MeOC6H4)Ti(O-i-Pr)3 (4e) S3 2. (2,6-Me2C6H3)Ti(O-i-Pr)3 (4j) S5 II. 1H and 13C NMR Spectra of Coupling Products S7 1. 1-Phenyl-1,2-propadiene (2aa) S7 2. 1-(4-Methylphenyl)-1,2-propadiene (2ab) S9 3. 1-(2-Methylphenyl)-1,2-propadiene (2ac) S11 4. 1-(4-Methoxyphenyl)-1,2-propadiene (2ad) S13 5. 1-(2-Methoxyphenyl)-1,2-propadiene (2ae) S15 6. 1-(3,5-Dimethylphenyl)-1,2-propadiene (2af) S17 7. 1-(2-Naphthyl)-1,2-propadiene (2ag) S19 8. 1-(4-Trifluoromethylphenyl)-1,2-propadiene (2ah) S21 9. 1-cyclohexyl-1,2-propadiene (2ai) S23 10. 1-(2,6-Dimethylphenyl)-1,2-propadiene (2aj) S25 11. 1-(2,6-Dimethylphenyl)-4-(bromomethyl)-1,2,4-pentatriene (2aj’) S27 12. 3-Phenyl-1,2-pentadiene (2ba) S29 13. 3-(2-Methylphenyl)-1,2-pentadiene (2bc) S31 14. 3-(3,5-Dimethylphenyl)-1,2-pentadiene (2bf) S33 15. 3-(2,6-Dimethylphenyl)-1,2-pentadiene (2bj) S35 16. -

House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, Or Isothiocyanate Radicals
House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals Agriculture Handbook No. 403 Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE Contents Page Methods 1 Results and discussion 3 Thiocyanic acid esters 8 Straight-chain nitriles 10 Propionitrile derivatives 10 Conclusions 24 Summary 25 Literature cited 26 This publication reports research involving pesticides. It does not contain recommendations for their use, nor does it imply that the uses discussed here have been registered. All uses of pesticides must be registered by appropriate State and Federal agencies before they can be recommended. CAUTION: Pesticides can be injurious to humans, domestic animals, desirable plants, and fish or other wildlife—if they are not handled or applied properly. Use all pesticides selectively and carefully. Follow recommended practices for the disposal of surplus pesticides and pesticide containers. ¿/áepé4áaUÁí^a¡eé —' ■ -"" TMK LABIL Mention of a proprietary product in this publication does not constitute a guarantee or warranty by the U.S. Department of Agriculture over other products not mentioned. Washington, D.C. Issued July 1971 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price 25 cents House Fly Attractants and Arrestants: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals BY M. S. MAYER, Entomology Research Division, Agricultural Research Service ^ Few chemicals possessing cyanide (-CN), thio- cyanate was slightly attractive to Musca domes- eyanate (-SCN), or isothiocyanate (~NCS) radi- tica, but it was considered to be one of the better cals have been tested as attractants for the house repellents for Phormia regina (Meigen). -

Diphosphine Complexes on Alpo4-Sepiolite Supports
catalysts Article Hydrogenation of α,β-Unsaturated Carbonyl Compounds over Covalently Heterogenized Ru(II) Diphosphine Complexes on AlPO4-Sepiolite Supports Verónica Caballero 1, Rafael Estevez 1, Diego Luna 1,* , Felipa M. Bautista 1 , Antonio A. Romero 1 , Laura Aguado-Deblas 1 , Jesús Hidalgo-Carrillo 1 and Isabel Romero 2 1 Departamento de Química Orgánica, Campus de Rabanales, Universidad de Córdoba, Ed. Marie Curie, 14014 Córdoba, Spain; [email protected] (V.C.); [email protected] (R.E.); [email protected] (F.M.B.); [email protected] (A.A.R.); [email protected] (L.A.-D.); [email protected] (J.H.-C.) 2 Departament de Química and Serveis Tècnics de Recerca, Universitat de Girona, C/M. Aurèlia Campmany, 69, E-17003 Girona, Spain; [email protected] * Correspondence: [email protected]; Tel.: +34-957212065 Abstract: In this work, the covalent immobilization of two ruthenium(II) complexes, II II i.e., [Ru Cl (bpea){(S)(-)(BINAP)}](BF4), 1, and [Ru Cl(bpea)(DPPE)](BF4), 2, where BINAP = 2,2’- bis(diphenylphosphino)-1,1’-binaphthyl and DPPE = 1,2-bis(diphenylphosphino)ethane, have been obtained (AlPO4-Sepiolite@1 and AlPO4-Sepiolite@2) by using a N-tridentate ligand N,N-bis-(2- pyridylmethyl)ethylamine (bpea), linked to an amorphous AlPO4-Sepiolite (20/80) inorganic support. Citation: Caballero, V.; Estevez, R.; This AlPO4-sepiolite support is able to immobilize the double amount of ruthenium complex (1.65%) Luna, D.; Bautista, F.M.; Romero, than the amorphous AlPO4 (0.89%). Both heterogenized complexes have been assessed as catalysts A.A.; Aguado-Deblas, L.; in the liquid phase hydrogenation of several substrates with carbonyl and/or olefinic double bonds Hidalgo-Carrillo, J.; Romero, I. -

Heterocyclic Chemistrychemistry
HeterocyclicHeterocyclic ChemistryChemistry Professor J. Stephen Clark Room C4-04 Email: [email protected] 2011 –2012 1 http://www.chem.gla.ac.uk/staff/stephenc/UndergraduateTeaching.html Recommended Reading • Heterocyclic Chemistry – J. A. Joule, K. Mills and G. F. Smith • Heterocyclic Chemistry (Oxford Primer Series) – T. Gilchrist • Aromatic Heterocyclic Chemistry – D. T. Davies 2 Course Summary Introduction • Definition of terms and classification of heterocycles • Functional group chemistry: imines, enamines, acetals, enols, and sulfur-containing groups Intermediates used for the construction of aromatic heterocycles • Synthesis of aromatic heterocycles • Carbon–heteroatom bond formation and choice of oxidation state • Examples of commonly used strategies for heterocycle synthesis Pyridines • General properties, electronic structure • Synthesis of pyridines • Electrophilic substitution of pyridines • Nucleophilic substitution of pyridines • Metallation of pyridines Pyridine derivatives • Structure and reactivity of oxy-pyridines, alkyl pyridines, pyridinium salts, and pyridine N-oxides Quinolines and isoquinolines • General properties and reactivity compared to pyridine • Electrophilic and nucleophilic substitution quinolines and isoquinolines 3 • General methods used for the synthesis of quinolines and isoquinolines Course Summary (cont) Five-membered aromatic heterocycles • General properties, structure and reactivity of pyrroles, furans and thiophenes • Methods and strategies for the synthesis of five-membered heteroaromatics -

Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation
No.171 No.171 ResearchResearch ArticleArticle Development of Novel Methodologies in Organic Synthesis Based on Ate Complex Formation Keiichi Hirano1* and Masanobu Uchiyama1,2* 1 Graduate School of Pharmaceutical Sciences, The University of Tokyo,7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan 2 Elements Chemistry Laboratory, RIKEN,2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan E-mail: [email protected]; [email protected] Abstract: We have been focusing on development of novel synthetic methodologies based on ate complex formation. By fine-tuning of coordination environments of various main-group metals (Zn, Al, and B) as well as transition metal (Cu), a wide range of carbon-carbon bond and carbon-heteroatom bond (C–I, C–O, C–N, C–H, C–Si, and C–B) formation reactions have been realized in highly regio- and chemoselective manner taking advantage of characteristics of the elements. Keywords: Ate complex, Halogen-metal exchange, Deprotonative metalation, Chemoselectivity, Regioselectivity 1. Introduction 2. Ate Complexes Ate complexes are known to be anionic organometallic Discovery of the first mono-anion type zincate, Na[ZnEt3], complexes, whose central metals have increased valence by by James A. Wanklyn dates back to 1859,1) and even di-anion accepting Lewis basic ligands to their vacant orbitals. They type zincate, Li2[ZnMe4], was already reported by Dallas T. are attractive chemical species offering tunable reactivity due Hurd in 1948.2) Zincates experienced a long blank after these to their flexible choices of central metals, counter cations, important findings before they started to attract attentions and coordination environments.