Systematic Expression Analysis of the Diagnostic and Prognostic Value of HEPACAM Family Member 2 in Colon Adenocarcinoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

5-Azacytidine Inhibits the Proliferation of Bladder Cancer Cells Via Reversal of the Aberrant Hypermethylation of the Hepacam Gene
ONCOLOGY REPORTS 35: 1375-1384, 2016 5-Azacytidine inhibits the proliferation of bladder cancer cells via reversal of the aberrant hypermethylation of the hepaCAM gene XIAORONG WANG1, E. CHEN1, XUE YANG1, YIN WANG1, ZHEN QUAN2, XIAOHOU WU2 and CHUNLI LUO1 1Key Laboratory of Diagnostic Medicine designated by the Chinese Ministry of Education, Chongqing Medical University; 2Department of Urology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, P.R. China Received June 14, 2015; Accepted July 24, 2015 DOI: 10.3892/or.2015.4492 Abstract. Hepatocyte cell adhesion molecule (hepaCAM), a cells. In addition, AZAC inhibited the proliferation of bladder tumor-suppressor gene, is rarely expressed in bladder carci- cancer cells and arrested cells at the G0/G1 phase. The in vivo noma. However, little is known concerning the mechanisms of results showed that expression of DNMT3A/3B and hepaCAM low hepaCAM expression in bladder cancer. Abnormal hyper- as well as tumor growth of nude mice were markedly altered methylation in the promoter plays a crucial role in cancer by which corresponded with the in vitro results. Due to the ability silencing tumor-suppressor genes, which is catalyzed by DNA to reactivate expression of hepaCAM and inhibit growth of methyltransferases (DNMTs). In the present study, a total of bladder cancer cells, AZAC may represent an effective treat- 31 bladder cancer and 22 adjacent tissues were assessed by ment for bladder cancer. immunohistochemistry to detect DNMT3A/3B and hepaCAM expression. Methylation of hepaCAM was determined by Introduction methylation‑specific polymerase chain reaction (MSP). The mRNA and protein levels of DNMT3A/3B and hepaCAM Hepatocyte cell adhesion molecule (hepaCAM), also known were determined by RT-PCR and western blot analysis after as GliaCAM, is located on chromosome 11q24 and encodes a treatment with 5-azacytidine (AZAC). -

Hepacam Inhibits Cell Proliferation and Invasion in Prostate Cancer by Suppressing Nuclear Translocation of the Androgen Receptor Via Its Cytoplasmic Domain
MOLECULAR MEDICINE REPORTS 19: 2115-2124, 2019 HepaCAM inhibits cell proliferation and invasion in prostate cancer by suppressing nuclear translocation of the androgen receptor via its cytoplasmic domain QINGFU DENG1, LI LUO2, ZHEN QUAN1, NANJING LIU2, ZHONGBO DU1, WEI SUN1, CHUNLI LUO2 and XIAOHOU WU1 1Department of Urology, First Affiliated Hospital of Chongqing Medical University;2 Key Laboratory of Diagnostics Medicine Designated by The Ministry of Education, Chongqing Medical University, Chongqing 400042, P.R. China Received May 23, 2018; Accepted December 12, 2018 DOI: 10.3892/mmr.2019.9841 Abstract. Hepatocyte cell adhesion molecule (HepaCAM) is other industrialized countries (1). Therefore, it is necessary to a tumour suppressor. However, the mechanism of HepaCAM elucidate the underlying pathophysiological processes of PCa function in prostate cancer (PCa) remains unknown. In with the advent of global ageing. the present study, HepaCAM, androgen receptor (AR) Hepatocyte cell adhesion molecule (HepaCAM) was first and Ran were analysed in 46 PCa tissue samples using detected in the liver (2), and it was later identified as a member immunohistochemistry. Subsequently, the influence of of the immunoglobulin superfamily. Members of the immu- HepaCAM and its cytoplasmic domain on cell proliferation, noglobulin superfamily are primarily localized at the cell migration, and invasion, and associated proteins was examined membrane and are composed of three domains: Extracellular, using MTT, wound healing, Transwell and western blotting transmembrane and cytoplasmic (3-5). The cytoplasmic assays, respectively. Furthermore, nuclear translocation of domain is fundamental to its biological function (2). Recent AR and Ran was analysed using immunofluorescence and studies have indicated that HepaCAM is present at low levels Western blot assays. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Genomics of Inherited Bone Marrow Failure and Myelodysplasia Michael
Genomics of inherited bone marrow failure and myelodysplasia Michael Yu Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2015 Reading Committee: Mary-Claire King, Chair Akiko Shimamura Marshall Horwitz Program Authorized to Offer Degree: Molecular and Cellular Biology 1 ©Copyright 2015 Michael Yu Zhang 2 University of Washington ABSTRACT Genomics of inherited bone marrow failure and myelodysplasia Michael Yu Zhang Chair of the Supervisory Committee: Professor Mary-Claire King Department of Medicine (Medical Genetics) and Genome Sciences Bone marrow failure and myelodysplastic syndromes (BMF/MDS) are disorders of impaired blood cell production with increased leukemia risk. BMF/MDS may be acquired or inherited, a distinction critical for treatment selection. Currently, diagnosis of these inherited syndromes is based on clinical history, family history, and laboratory studies, which directs the ordering of genetic tests on a gene-by-gene basis. However, despite extensive clinical workup and serial genetic testing, many cases remain unexplained. We sought to define the genetic etiology and pathophysiology of unclassified bone marrow failure and myelodysplastic syndromes. First, to determine the extent to which patients remained undiagnosed due to atypical or cryptic presentations of known inherited BMF/MDS, we developed a massively-parallel, next- generation DNA sequencing assay to simultaneously screen for mutations in 85 BMF/MDS genes. Querying 71 pediatric and adult patients with unclassified BMF/MDS using this assay revealed 8 (11%) patients with constitutional, pathogenic mutations in GATA2 , RUNX1 , DKC1 , or LIG4 . All eight patients lacked classic features or laboratory findings for their syndromes. -

Somatic Mutations and Progressive Monosomy Modify SAMD9-Related Phenotypes in Humans
Downloaded from http://www.jci.org on June 26, 2017. https://doi.org/10.1172/JCI91913 RESEARCH ARTICLE The Journal of Clinical Investigation Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans Federica Buonocore,1 Peter Kühnen,2 Jenifer P. Suntharalingham,1 Ignacio Del Valle,1 Martin Digweed,3 Harald Stachelscheid,4 Noushafarin Khajavi,2 Mohammed Didi,5 Angela F. Brady,6 Oliver Blankenstein,2 Annie M. Procter,7 Paul Dimitri,8 Jerry K.H. Wales,9 Paolo Ghirri,10 Dieter Knöbl,11 Brigitte Strahm,12 Miriam Erlacher,12,13 Marcin W. Wlodarski,12,13 Wei Chen,14 George K. Kokai,15 Glenn Anderson,16 Deborah Morrogh,17 Dale A. Moulding,18 Shane A. McKee,19 Charlotte M. Niemeyer,12,13 Annette Grüters,2 and John C. Achermann1 1Genetics and Genomic Medicine, University College London (UCL) Great Ormond Street Institute of Child Health, London, United Kingdom. 2Institute of Experimental Pediatric Endocrinology and Department of Pediatric Endocrinology, Charité, Berlin, Germany. 3Department of Human and Medical Genetics, Charité, Berlin, Germany. 4Berlin Institute of Health, Berlin, Germany, and Berlin-Brandenburg Centrum for Regenerative Therapies, Charité, Berlin, Germany. 5Department of Paediatric Endocrinology, Alder Hey Children’s NHS Foundation Trust, Liverpool, United Kingdom. 6North West Thames Regional Genetics Service, Northwick Park Hospital, Harrow, United Kingdom. 7Institute of Medical Genetics, University Hospital of Wales, Cardiff, United Kingdom. 8Academic Unit of Child Health, University of Sheffield, Sheffield, United Kingdom. 9Department of Endocrinology, Children’s Health Queensland Clinical Unit, University of Queensland, Brisbane, Australia. 10Department of Neonatology, University of Pisa, Pisa, Italy. 11Pediatric Endocrinology, Karlsruhe, Germany. -

Of Small Intestine Harboring Driver Gene Mutations: a Case Report and a Literature Review
1161 Case Report A rare multiple primary sarcomatoid carcinoma (SCA) of small intestine harboring driver gene mutations: a case report and a literature review Zhu Zhu1#, Xinyi Liu2#, Wenliang Li1, Zhengqi Wen1, Xiang Ji1, Ruize Zhou1, Xiaoyu Tuo3, Yaru Chen2, Xian Gong2, Guifeng Liu2, Yanqing Zhou2, Shifu Chen2, Lele Song2#^, Jian Huang1 1Department of Oncology, First Affiliated Hospital of Kunming Medical University, Kunming, China; 2HaploX Biotechnology, Shenzhen, China; 3Department of Pathology, First Affiliated Hospital of Kunming Medical University, Kunming, China #These authors contributed equally to this work. Correspondence to: Jian Huang. Department of Oncology, First Affiliated Hospital of Kunming Medical University, No. 295, Xichang Road, Kunming 560032, Yunnan Province, China. Email: [email protected]; Lele Song. HaploX Biotechnology, 8th floor, Auto Electric Power Building, Songpingshan Road, Nanshan District, Shenzhen 518057, Guangdong Province, China. Email: [email protected]. Abstract: Primary sarcomatoid carcinoma (SCA) is a type of rare tumor consisting of both malignant epithelial and mesenchymal components. Only 32 cases of SCA of the small bowel have been reported in the literature to date. Due to its rarity and complexity, this cancer has not been genetically studied and its diagnosis and treatment remain difficult. Here we report a 54-year-old male underwent emergency surgical resection in the small intestine due to severe obstruction and was diagnosed with multiple SCA based on postoperative pathological examination. Over 100 polypoid tumors scattered along his whole jejunum and proximal ileum. Chemotherapy (IFO+Epirubicin) was performed after surgery while the patient died two months after the surgery due to severe malnutrition. Whole-exome sequencing was performed for the tumor tissue with normal tissue as the control. -

3,5-T3 in Brain and Liver Uncovers Novel Roles for Thyroid
www.nature.com/scientificreports OPEN Diferential transcriptome regulation by 3,5-T2 and 3′,3,5-T3 in brain and liver uncovers novel Received: 25 May 2017 Accepted: 16 October 2017 roles for thyroid hormones in tilapia Published: xx xx xxxx A. Olvera1, C. J. Martyniuk2, N. Buisine3, V. Jiménez-Jacinto4, A. Sanchez-Flores4, L. M. Sachs3 & A. Orozco 1 Although 3,5,3′-triiodothyronine (T3) is considered to be the primary bioactive thyroid hormone (TH) due to its high afnity for TH nuclear receptors (TRs), new data suggest that 3,5-diiodothyronine (T2) can also regulate transcriptional networks. To determine the functional relevance of these bioactive THs, RNA-seq analysis was conducted in the cerebellum, thalamus-pituitary and liver of tilapia treated with equimolar doses of T2 or T3. We identifed a total of 169, 154 and 2863 genes that were TH- responsive (FDR < 0.05) in the tilapia cerebellum, thalamus-pituitary and liver, respectively. Among these, 130, 96 and 349 genes were uniquely regulated by T3, whereas 22, 40 and 929 were exclusively regulated by T2 under our experimental paradigm. The expression profles in response to TH treatment were tissue-specifc, and the diversity of regulated genes also resulted in a variety of diferent pathways being afected by T2 and T3. T2 regulated gene networks associated with cell signalling and transcriptional pathways, while T3 regulated pathways related to cell signalling, the immune system, and lipid metabolism. Overall, the present work highlights the relevance of T2 as a key bioactive hormone, and reveals some of the diferent functional strategies that underpin TH pleiotropy. -
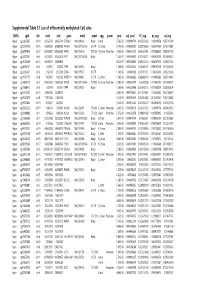
Supplementary Table S1 Kim Et Al.Xlsx
Supplemental Table S1. List of differentially methylated CpG sites DMRs cgid chr start end gene nmid annot cpg prom pval adj. pval FC_avg nc_avg cd_avg Hyper cg12827637 chr14 69256790 69256791 ZFP36L1 NM_004926 Body Island . 5.76E-05 0.000989714 4.465355492 0.06419878 0.287016909 Hyper cg02072495 chr15 60689284 60689285 ANXA2 NM_001136015 5'UTR N_Shore . 2.04E-05 0.000560282 3.808196653 0.056572464 0.215719887 Hyper cg26147845 chr12 132433837 132433838 EP400 NM_015409 TSS1500N_Shore Promoter 3.92E-06 0.000301217 3.643351981 0.077868629 0.283967159 Hyper cg23737061 chr3 60226327 60226328 FHIT NM_001166243 Body . 1.65E-07 0.000239667 3.616168212 0.093920309 0.339893251 Hyper cg18215449 chr12 66089472 66089473 . 4.22E-07 0.000239667 3.565225234 0.064067475 0.228671502 Hyper cg24082121 chr5 672871 672872 TPPP NM_007030 Body . 1.78E-05 0.000525064 3.505843071 0.089095718 0.312606189 Hyper cg23032421 chr3 3152037 3152038 IL5RA NM_175727 5'UTR . 1.04E-05 0.00041338 3.371418731 0.114213604 0.385299026 Hyper cg11737172 chr8 1923051 1923052 KBTBD11 NM_014867 5'UTR S_Shore . 1.38E-05 0.000466865 3.368483202 0.108495063 0.365700645 Hyper cg16886175 chr7 114562062 114562063 MDFIC NM_001166345 TSS200 N_Shore Promoter 7.28E-07 0.000243374 3.35638256 0.101463003 0.340784291 Hyper cg10986412 chr5 672909 672910 TPPP NM_007030 Body . 1.56E-06 0.000255546 3.316242102 0.071981379 0.238939304 Hyper cg02025737 chr15 33384750 33384751 . 3.80E-05 0.000776402 3.312390381 0.13643852 0.452168879 Hyper cg06243675 chr8 11801364 11801365 . 5.23E-05 0.000932639 3.254014485 0.016251643 0.053108482 Hyper cg09577804 chr10 3235531 3235532 . 5.04E-05 0.000912242 3.247042327 0.043496295 0.141459016 Hyper cg23057220 chr19 1356314 1356315 MUM1 NR_024247 TSS200 S_Shore Promoter 3.65E-05 0.000758178 3.204370027 0.14549078 0.466426732 Hyper cg06984883 chr1 1243563 1243564 PUSL1 NM_153339 TSS1500Island Promoter 5.07E-06 0.000323878 3.198831858 0.005047892 0.01636724 Hyper cg15963463 chr1 25253236 25253237 RUNX3 NM_001031680 Body N_Shelf . -

Somatic Mutations and Progressive Monosomy Modify SAMD9-Related Phenotypes in Humans
Downloaded from http://www.jci.org on June 5, 2017. https://doi.org/10.1172/JCI91913 RESEARCH ARTICLE The Journal of Clinical Investigation Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans Federica Buonocore,1 Peter Kühnen,2 Jenifer P. Suntharalingham,1 Ignacio Del Valle,1 Martin Digweed,3 Harald Stachelscheid,4 Noushafarin Khajavi,2 Mohammed Didi,5 Angela F. Brady,6 Oliver Blankenstein,2 Annie M. Procter,7 Paul Dimitri,8 Jerry K.H. Wales,9 Paolo Ghirri,10 Dieter Knöbl,11 Brigitte Strahm,12 Miriam Erlacher,12,13 Marcin W. Wlodarski,12,13 Wei Chen,14 George K. Kokai,15 Glenn Anderson,16 Deborah Morrogh,17 Dale A. Moulding,18 Shane A. McKee,19 Charlotte M. Niemeyer,12,13 Annette Grüters,2 and John C. Achermann1 1Genetics and Genomic Medicine, University College London (UCL) Great Ormond Street Institute of Child Health, London, United Kingdom. 2Institute of Experimental Pediatric Endocrinology and Department of Pediatric Endocrinology, Charité, Berlin, Germany. 3Department of Human and Medical Genetics, Charité, Berlin, Germany. 4Berlin Institute of Health, Berlin, Germany, and Berlin-Brandenburg Centrum for Regenerative Therapies, Charité, Berlin, Germany. 5Department of Paediatric Endocrinology, Alder Hey Children’s NHS Foundation Trust, Liverpool, United Kingdom. 6North West Thames Regional Genetics Service, Northwick Park Hospital, Harrow, United Kingdom. 7Institute of Medical Genetics, University Hospital of Wales, Cardiff, United Kingdom. 8Academic Unit of Child Health, University of Sheffield, Sheffield, United Kingdom. 9Department of Endocrinology, Children’s Health Queensland Clinical Unit, University of Queensland, Brisbane, Australia. 10Department of Neonatology, University of Pisa, Pisa, Italy. 11Pediatric Endocrinology, Karlsruhe, Germany. -

Genome-Wide Association Study for Feed Efficiency and Growth Traits in U.S. Beef Cattle Christopher M
Seabury et al. BMC Genomics (2017) 18:386 DOI 10.1186/s12864-017-3754-y RESEARCH ARTICLE Open Access Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle Christopher M. Seabury1*, David L. Oldeschulte1, Mahdi Saatchi2, Jonathan E. Beever3, Jared E. Decker4,5, Yvette A. Halley1, Eric K. Bhattarai1, Maral Molaei1, Harvey C. Freetly6, Stephanie L. Hansen2, Helen Yampara-Iquise4, Kristen A. Johnson7, Monty S. Kerley4, JaeWoo Kim4, Daniel D. Loy2, Elisa Marques8, Holly L. Neibergs7, Robert D. Schnabel4,5, Daniel W. Shike3, Matthew L. Spangler9, Robert L. Weaber10, Dorian J. Garrick2,11 and Jeremy F. Taylor4* Abstract Background: Single nucleotide polymorphism (SNP) arrays for domestic cattle have catalyzed the identification of genetic markers associated with complex traits for inclusion in modern breeding and selection programs. Using actual and imputed Illumina 778K genotypes for 3887 U.S. beef cattle from 3 populations (Angus, Hereford, SimAngus), we performed genome-wide association analyses for feed efficiency and growth traits including average daily gain (ADG), dry matter intake (DMI), mid-test metabolic weight (MMWT), and residual feed intake (RFI), with marker-based heritability estimates produced for all traits and populations. Results: Moderate and/or large-effect QTL were detected for all traits in all populations, as jointly defined by the estimated proportion of variance explained (PVE) by marker effects (PVE ≥ 1.0%) and a nominal P-value threshold (P ≤ 5e-05). Lead SNPs with PVE ≥ 2.0% were considered putative evidence of large-effect QTL (n = 52), whereas those with PVE ≥ 1.0% but < 2.0% were considered putative evidence for moderate-effect QTL (n = 35). -

Early Mechanisms of Retinal Degeneration in the Harlequin Mouse
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 7-17-2014 12:00 AM Early Mechanisms of Retinal Degeneration in the harlequin Mouse Eric Dolinar The University of Western Ontario Supervisor Dr. Kathleen Hill The University of Western Ontario Graduate Program in Biology A thesis submitted in partial fulfillment of the equirr ements for the degree in Master of Science © Eric Dolinar 2014 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Biology Commons, Genetics Commons, and the Molecular Genetics Commons Recommended Citation Dolinar, Eric, "Early Mechanisms of Retinal Degeneration in the harlequin Mouse" (2014). Electronic Thesis and Dissertation Repository. 2156. https://ir.lib.uwo.ca/etd/2156 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. i Early Mechanisms of Retinal Degeneration in the harlequin Mouse (Thesis Format: Monograph) by Eric A Dolinar Graduate Program in Biology A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science The School of Graduate and Postdoctoral Studies The University of Western Ontario, London, Ontario, Canada © Eric A Dolinar 2014 i ii Abstract Retinal diseases are personally debilitating and expensive, yet many early disease mechanisms leading to their onset and progression remain poorly understood. The harlequin mouse is a model of human mitochondrial dysfunction and parainflammation leading to subsequent cerebellar and retinal degeneration. Diagnosis of retinal degeneration can be tracked in vivo and is associated with AIF dysfunction.