Hepacam Associates with Connexin 43 and Enhances Its Localization In
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

5-Azacytidine Inhibits the Proliferation of Bladder Cancer Cells Via Reversal of the Aberrant Hypermethylation of the Hepacam Gene
ONCOLOGY REPORTS 35: 1375-1384, 2016 5-Azacytidine inhibits the proliferation of bladder cancer cells via reversal of the aberrant hypermethylation of the hepaCAM gene XIAORONG WANG1, E. CHEN1, XUE YANG1, YIN WANG1, ZHEN QUAN2, XIAOHOU WU2 and CHUNLI LUO1 1Key Laboratory of Diagnostic Medicine designated by the Chinese Ministry of Education, Chongqing Medical University; 2Department of Urology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, P.R. China Received June 14, 2015; Accepted July 24, 2015 DOI: 10.3892/or.2015.4492 Abstract. Hepatocyte cell adhesion molecule (hepaCAM), a cells. In addition, AZAC inhibited the proliferation of bladder tumor-suppressor gene, is rarely expressed in bladder carci- cancer cells and arrested cells at the G0/G1 phase. The in vivo noma. However, little is known concerning the mechanisms of results showed that expression of DNMT3A/3B and hepaCAM low hepaCAM expression in bladder cancer. Abnormal hyper- as well as tumor growth of nude mice were markedly altered methylation in the promoter plays a crucial role in cancer by which corresponded with the in vitro results. Due to the ability silencing tumor-suppressor genes, which is catalyzed by DNA to reactivate expression of hepaCAM and inhibit growth of methyltransferases (DNMTs). In the present study, a total of bladder cancer cells, AZAC may represent an effective treat- 31 bladder cancer and 22 adjacent tissues were assessed by ment for bladder cancer. immunohistochemistry to detect DNMT3A/3B and hepaCAM expression. Methylation of hepaCAM was determined by Introduction methylation‑specific polymerase chain reaction (MSP). The mRNA and protein levels of DNMT3A/3B and hepaCAM Hepatocyte cell adhesion molecule (hepaCAM), also known were determined by RT-PCR and western blot analysis after as GliaCAM, is located on chromosome 11q24 and encodes a treatment with 5-azacytidine (AZAC). -

Hepacam Inhibits Cell Proliferation and Invasion in Prostate Cancer by Suppressing Nuclear Translocation of the Androgen Receptor Via Its Cytoplasmic Domain
MOLECULAR MEDICINE REPORTS 19: 2115-2124, 2019 HepaCAM inhibits cell proliferation and invasion in prostate cancer by suppressing nuclear translocation of the androgen receptor via its cytoplasmic domain QINGFU DENG1, LI LUO2, ZHEN QUAN1, NANJING LIU2, ZHONGBO DU1, WEI SUN1, CHUNLI LUO2 and XIAOHOU WU1 1Department of Urology, First Affiliated Hospital of Chongqing Medical University;2 Key Laboratory of Diagnostics Medicine Designated by The Ministry of Education, Chongqing Medical University, Chongqing 400042, P.R. China Received May 23, 2018; Accepted December 12, 2018 DOI: 10.3892/mmr.2019.9841 Abstract. Hepatocyte cell adhesion molecule (HepaCAM) is other industrialized countries (1). Therefore, it is necessary to a tumour suppressor. However, the mechanism of HepaCAM elucidate the underlying pathophysiological processes of PCa function in prostate cancer (PCa) remains unknown. In with the advent of global ageing. the present study, HepaCAM, androgen receptor (AR) Hepatocyte cell adhesion molecule (HepaCAM) was first and Ran were analysed in 46 PCa tissue samples using detected in the liver (2), and it was later identified as a member immunohistochemistry. Subsequently, the influence of of the immunoglobulin superfamily. Members of the immu- HepaCAM and its cytoplasmic domain on cell proliferation, noglobulin superfamily are primarily localized at the cell migration, and invasion, and associated proteins was examined membrane and are composed of three domains: Extracellular, using MTT, wound healing, Transwell and western blotting transmembrane and cytoplasmic (3-5). The cytoplasmic assays, respectively. Furthermore, nuclear translocation of domain is fundamental to its biological function (2). Recent AR and Ran was analysed using immunofluorescence and studies have indicated that HepaCAM is present at low levels Western blot assays. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

3,5-T3 in Brain and Liver Uncovers Novel Roles for Thyroid
www.nature.com/scientificreports OPEN Diferential transcriptome regulation by 3,5-T2 and 3′,3,5-T3 in brain and liver uncovers novel Received: 25 May 2017 Accepted: 16 October 2017 roles for thyroid hormones in tilapia Published: xx xx xxxx A. Olvera1, C. J. Martyniuk2, N. Buisine3, V. Jiménez-Jacinto4, A. Sanchez-Flores4, L. M. Sachs3 & A. Orozco 1 Although 3,5,3′-triiodothyronine (T3) is considered to be the primary bioactive thyroid hormone (TH) due to its high afnity for TH nuclear receptors (TRs), new data suggest that 3,5-diiodothyronine (T2) can also regulate transcriptional networks. To determine the functional relevance of these bioactive THs, RNA-seq analysis was conducted in the cerebellum, thalamus-pituitary and liver of tilapia treated with equimolar doses of T2 or T3. We identifed a total of 169, 154 and 2863 genes that were TH- responsive (FDR < 0.05) in the tilapia cerebellum, thalamus-pituitary and liver, respectively. Among these, 130, 96 and 349 genes were uniquely regulated by T3, whereas 22, 40 and 929 were exclusively regulated by T2 under our experimental paradigm. The expression profles in response to TH treatment were tissue-specifc, and the diversity of regulated genes also resulted in a variety of diferent pathways being afected by T2 and T3. T2 regulated gene networks associated with cell signalling and transcriptional pathways, while T3 regulated pathways related to cell signalling, the immune system, and lipid metabolism. Overall, the present work highlights the relevance of T2 as a key bioactive hormone, and reveals some of the diferent functional strategies that underpin TH pleiotropy. -
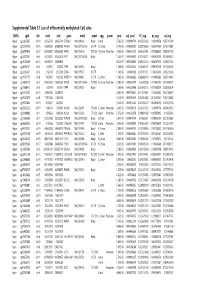
Supplementary Table S1 Kim Et Al.Xlsx
Supplemental Table S1. List of differentially methylated CpG sites DMRs cgid chr start end gene nmid annot cpg prom pval adj. pval FC_avg nc_avg cd_avg Hyper cg12827637 chr14 69256790 69256791 ZFP36L1 NM_004926 Body Island . 5.76E-05 0.000989714 4.465355492 0.06419878 0.287016909 Hyper cg02072495 chr15 60689284 60689285 ANXA2 NM_001136015 5'UTR N_Shore . 2.04E-05 0.000560282 3.808196653 0.056572464 0.215719887 Hyper cg26147845 chr12 132433837 132433838 EP400 NM_015409 TSS1500N_Shore Promoter 3.92E-06 0.000301217 3.643351981 0.077868629 0.283967159 Hyper cg23737061 chr3 60226327 60226328 FHIT NM_001166243 Body . 1.65E-07 0.000239667 3.616168212 0.093920309 0.339893251 Hyper cg18215449 chr12 66089472 66089473 . 4.22E-07 0.000239667 3.565225234 0.064067475 0.228671502 Hyper cg24082121 chr5 672871 672872 TPPP NM_007030 Body . 1.78E-05 0.000525064 3.505843071 0.089095718 0.312606189 Hyper cg23032421 chr3 3152037 3152038 IL5RA NM_175727 5'UTR . 1.04E-05 0.00041338 3.371418731 0.114213604 0.385299026 Hyper cg11737172 chr8 1923051 1923052 KBTBD11 NM_014867 5'UTR S_Shore . 1.38E-05 0.000466865 3.368483202 0.108495063 0.365700645 Hyper cg16886175 chr7 114562062 114562063 MDFIC NM_001166345 TSS200 N_Shore Promoter 7.28E-07 0.000243374 3.35638256 0.101463003 0.340784291 Hyper cg10986412 chr5 672909 672910 TPPP NM_007030 Body . 1.56E-06 0.000255546 3.316242102 0.071981379 0.238939304 Hyper cg02025737 chr15 33384750 33384751 . 3.80E-05 0.000776402 3.312390381 0.13643852 0.452168879 Hyper cg06243675 chr8 11801364 11801365 . 5.23E-05 0.000932639 3.254014485 0.016251643 0.053108482 Hyper cg09577804 chr10 3235531 3235532 . 5.04E-05 0.000912242 3.247042327 0.043496295 0.141459016 Hyper cg23057220 chr19 1356314 1356315 MUM1 NR_024247 TSS200 S_Shore Promoter 3.65E-05 0.000758178 3.204370027 0.14549078 0.466426732 Hyper cg06984883 chr1 1243563 1243564 PUSL1 NM_153339 TSS1500Island Promoter 5.07E-06 0.000323878 3.198831858 0.005047892 0.01636724 Hyper cg15963463 chr1 25253236 25253237 RUNX3 NM_001031680 Body N_Shelf . -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -

Molecular Sciences High-Resolution Chromosome Ideogram Representation of Currently Recognized Genes for Autism Spectrum Disorder
Int. J. Mol. Sci. 2015, 16, 6464-6495; doi:10.3390/ijms16036464 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article High-Resolution Chromosome Ideogram Representation of Currently Recognized Genes for Autism Spectrum Disorders Merlin G. Butler *, Syed K. Rafi † and Ann M. Manzardo † Departments of Psychiatry & Behavioral Sciences and Pediatrics, University of Kansas Medical Center, Kansas City, KS 66160, USA; E-Mails: [email protected] (S.K.R.); [email protected] (A.M.M.) † These authors contributed to this work equally. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-913-588-1873; Fax: +1-913-588-1305. Academic Editor: William Chi-shing Cho Received: 23 January 2015 / Accepted: 16 March 2015 / Published: 20 March 2015 Abstract: Recently, autism-related research has focused on the identification of various genes and disturbed pathways causing the genetically heterogeneous group of autism spectrum disorders (ASD). The list of autism-related genes has significantly increased due to better awareness with advances in genetic technology and expanding searchable genomic databases. We compiled a master list of known and clinically relevant autism spectrum disorder genes identified with supporting evidence from peer-reviewed medical literature sources by searching key words related to autism and genetics and from authoritative autism-related public access websites, such as the Simons Foundation Autism Research Institute autism genomic database dedicated to gene discovery and characterization. Our list consists of 792 genes arranged in alphabetical order in tabular form with gene symbols placed on high-resolution human chromosome ideograms, thereby enabling clinical and laboratory geneticists and genetic counsellors to access convenient visual images of the location and distribution of ASD genes. -
Mechanism of Fibrosis in HNF1B-Related Autosomal Dominant Tubulointerstitial Kidney Disease
BASIC RESEARCH www.jasn.org Mechanism of Fibrosis in HNF1B-Related Autosomal Dominant Tubulointerstitial Kidney Disease Siu Chiu Chan,1 Ying Zhang,2 Annie Shao,1 Svetlana Avdulov,1 Jeremy Herrera,1 Karam Aboudehen,1 Marco Pontoglio,3 and Peter Igarashi 1 1Department of Medicine and 2Minnesota Supercomputing Institute, University of Minnesota, Minneapolis, Minnesota; and 3Department of Development, Reproduction and Cancer, Institut Cochin, Institut National de la Santé et de la Recherche Médicale U1016/Centre National de la Recherche Scientifique Unité Mixte de Recherche 8104, Université Paris-Descartes, Paris, France ABSTRACT Background Mutation of HNF1B, the gene encoding transcription factor HNF-1b, is one cause of auto- somal dominant tubulointerstitial kidney disease, a syndrome characterized by tubular cysts, renal fibrosis, and progressive decline in renal function. HNF-1b has also been implicated in epithelial–mesenchymal transition (EMT) pathways, and sustained EMT is associated with tissue fibrosis. The mechanism whereby mutated HNF1B leads to tubulointerstitial fibrosis is not known. Methods To explore the mechanism of fibrosis, we created HNF-1b–deficient mIMCD3 renal epithelial cells, used RNA-sequencing analysis to reveal differentially expressed genes in wild-type and HNF-1b–deficient mIMCD3 cells, and performed cell lineage analysis in HNF-1b mutant mice. Results The HNF-1b–deficient cells exhibited properties characteristic of mesenchymal cells such as fi- broblasts, including spindle-shaped morphology, loss of contact inhibition, and increased cell migration. These cells also showed upregulation of fibrosis and EMT pathways, including upregulation of Twist2, Snail1, Snail2, and Zeb2, which are key EMT transcription factors. Mechanistically, HNF-1b directly re- presses Twist2, and ablation of Twist2 partially rescued the fibroblastic phenotype of HNF-1b mutant cells. -

Systematic Expression Analysis of the Diagnostic and Prognostic Value of HEPACAM Family Member 2 in Colon Adenocarcinoma
Systematic Expression Analysis of the Diagnostic and Prognostic Value of HEPACAM Family Member 2 in Colon Adenocarcinoma Shuai Wang Guangxi Medical University First Aliated Hospital Guo-Tian Ruan Guangxi Medical University First Aliated Hospital Yi-Zhen Gong Guilin Medical University Aliated Hospital Cun Liao Guangxi Medical University First Aliated Hospital Lei Zhang Guilin Medical University Aliated Hospital Feng Gao ( [email protected] ) Guangxi Medical University First Aliated Hospital https://orcid.org/0000-0002-1000-385X Research article Keywords: HEPACAM2, Guangxi cohort, COAD, prognosis, diagnosis Posted Date: January 29th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-154487/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/19 Abstract Background: The diagnostic and prognostic value of HEPACAM family member 2 (HEPACAM2) gene in patients with colon adenocarcinoma (COAD) is rarely reported. Therefore, the purpose of this study is to explore the diagnostic and prognostic value of HEPACAM2 gene in patients with COAD. Methods: Firstly, we analyzed the differential expression levels of HEPACAM2 gene and diagnostic value analysis from different databases. Secondly, univariate and multivariate survival analysis of the prognostic value of HEPACAM2 gene in patients with COAD was performed. Finally, utilizing joint-effects analysis and comprehensive prognosis analysis to investigate the prognostic value of HEPACAM2 and related genes. Results: Differential analyses of multiple databases showed that the HEPACAM2 expression level in COAD tumor tissue was signicantly lower than that of adjacent normal tissues. The diagnostic ROC curve results indicated that HEPACAM2 gene had a higher diagnostic value in COAD. -

Investigations of the New Cell Adhesion Molecule Hepacam: Its Connection with Integrin-Dependent Signaling and Its Proteolytic Cleavage
INVESTIGATIONS OF THE NEW CELL ADHESION MOLECULE HEPACAM: ITS CONNECTION WITH INTEGRIN-DEPENDENT SIGNALING AND ITS PROTEOLYTIC CLEAVAGE ZHANG TING (B.Sc. (Hons.), National University of Singapore) A THESIS SUBMITTED FOR THE DEGREE OF DOCTOR OF PHILOSOPHY DEPARTMENT OF PHYSIOLOGY NATIONAL UNIVERSITY OF SINGAPORE 2011 ACKNOWLEDGEMENTS I would like to express my deepest gratefulness to my supervisor Dr. Shen Shali. Undoubtedly, Dr. Shen is the one who gave me the greatest influence during the past few years. I joined her lab as a UROPS student and stayed on for about 7 years because the group has such a wonderful leader. Dr. Shen has a great personal charm. From her, I learnt not only technical skills and knowledge, but also the attitude towards research and life. She makes me believe that real science must be motivated by genuine passion, and one must have some belief to hold in his/her life. To me, Dr. Shen is not only my supervisor, but also a friend for a lifetime. My sincere appreciations go to Prof Herbert Schwarz. He gave me many valuable advices on paper and thesis writing as well as tips for choosing a job. I have been deeply impressed by his seriousness and high efficiency at work. His generous help and kindness made me feel warm and calm during the hectic period before graduation. My warmest thanks go to my colleagues Dr. Angela Moh, Lee Lay Hoon and Zhang Chunli. I’m so fortunate to have them as lab mates and friends. They gave me tremendous help, teaching me all the technical skills and were always willing to give out a helping hand when I encountered difficulties. -

Cloning and Characterization of Hepacam, a Novel Ig-Like Cell Adhesion Molecule Suppressed in Human Hepatocellular Carcinoma
Journal of Hepatology 42 (2005) 833–841 www.elsevier.com/locate/jhep Cloning and characterization of hepaCAM, a novel Ig-like cell adhesion molecule suppressed in human hepatocellular carcinoma Mei Chung Moh, Lay Hoon Lee, Shali Shen* Laboratory of Hepato-Oncogenetics, Department of Physiology, Faculty of Medicine, National University of Singapore, 2 Medical Drive, Singapore 117597 Background/Aims: Previously, we reported on gene HEPN1 that was silenced in hepatocellular carcinoma (HCC) and its capability of arresting cell growth. In this study, we identified another novel gene hepaCAM from the liver, which contains the full-length HEPN1 on its antisense strand in the 30-noncoding region, and assessed its expression, characteristics and functions in HCC. Methods: Full-length hepaCAM cDNA was isolated by rapid amplification of cDNA ends. The gene expression was examined by semi-quantitative RT-PCR in 23 paired HCC liver specimens and 5 HCC cell lines. Transfection studies, coupled with immunocytochemistry, cellular interaction analyses, colony formation and microtetrazolium assay, were employed to elucidate the localization and functions of hepaCAM. Results: The expression of hepaCAM decreased in 20/23 of HCC samples and was undetectable in 5 HCC cell lines tested. The gene product consisting of 416 amino acids displayed the typical structure of Ig-like cell adhesion molecules. The protein was glycosylated and predominantly localized on the cytoplasmic membrane. When re-expressed in HepG2, hepaCAM accelerated cell spreading (P!0.001), increased cell motility (PZ0.0011), reduced colony formation (PZ0.0022), and inhibited cell growth (P!0.001). Conclusions: Gene hepaCAM, frequently silenced in HCC, encodes an Ig-like transmembrane glycoprotein and is involved in cell adhesion and growth control.